
acqua
Composto chimico di formula H2O, assai diffuso in natura nei suoi tre stati d’aggregazione: solido, liquido e aeriforme. Nel linguaggio corrente s’intende in genere l’a. allo stato liquido.
Chimica
Generalità
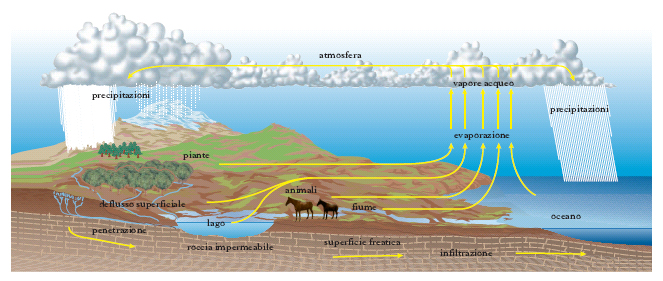
Per la sua abbondanza sulla superficie terrestre e negli organismi viventi gli antichi considerarono l’a. uno dei quattro elementi ed elemento l’a. fu ritenuta finché H. Cavendish (1781) e A.-L. Lavoisier (1783) ne stabilirono la composizione e il metodo di sintesi. L’a. si forma per mezzo di numerose reazioni (per azione dell’idrogeno sui composti ossigenati, per combustione dell’idrogeno o di composti contenenti idrogeno ecc.), ma normalmente non si prepara, data la diffusione con cui si presenta in natura in tutti e tre gli stati di aggregazione. Questi rappresentano anche le varie fasi di un ciclo chiuso (fig. 1) cui l’a. è sottoposta per azione del calore solare: dalla superficie marina o terrestre l’a. sale per evaporazione nell’atmosfera, ove si condensa e ricade sulla terra sotto forma di pioggia, neve, grandine, brina e rugiada; di essa parte resta in superficie, parte, attraverso fori e spaccature delle rocce, scende in zone più o meno profonde dove, arrestata dall’incontro di terreni impermeabili, scorre con leggi analoghe a quelle delle a. superficiali finché, per via naturale o per richiamo provocato artificialmente, ritorna in superficie, ove ricomincia il ciclo. Non tutta l’a. che arriva al suolo ritorna in circolazione, perché una certa porzione reagisce chimicamente con altre sostanze e non ricompare come a. finché nuovi processi geochimici non la liberano dai minerali nei quali è combinata; la perdita è compensata anche dalle a. di nuova formazione che si originano nelle grandi profondità per combinazione di idrogeno e ossigeno ad alta temperatura ( a. giovanili). Nel compiersi di tutto il ciclo idrologico l’a. viene a contatto con sostanze solide (soprattutto sali), liquide e gassose, che in parte dissolve o porta in sospensione; essa non è quindi mai pura, e ciò vale anche per l’a. meteorica che, attraversando strati più o meno spessi dell’atmosfera, porta in soluzione alcuni dei componenti dell’aria e trascina il pulviscolo atmosferico. Quando l’a. contiene disciolta dell’aria e talora anche altri gas, si dice aerata; quando vi sia stata eliminata (per es. attraverso ebollizione) l’aria normalmente discioltavi, o anche gli altri gas, si dice disaerata. Per ottenere l’a. allo stato puro bisogna sottoporla a ripetute distillazioni (in presenza anche di sostanze come permanganato di potassio, ossido di bario ecc., avendo cura d’impedire il disciogliersi di gas, come ossigeno, azoto, anidride carbonica ecc.).
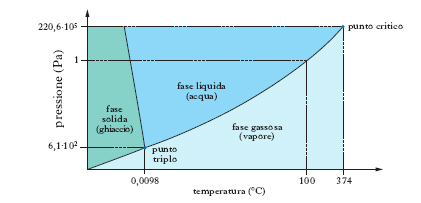
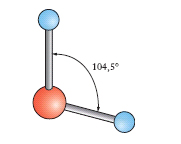
Nelle normali condizioni di temperatura e pressione l’a. si presenta sotto forma di un liquido limpido, inodore, insapore, incolore in strato sottile, ma che appare colorato in azzurro se osservato sotto spessore di qualche metro (causa il suo potere assorbente verso le radiazioni complementari di questo colore). Nella fig. 2 è indicato il diagramma d’equilibrio delle varie fasi dell’acqua. Nel punto O (punto triplo), cui corrisponde la temperatura di 0,0098 °C e la pressione di 610 Pa (tensione della fase vapore), coesistono le tre fasi: solido, liquido e vapore; le linee piene indicano l’equilibrio fra le fasi segnate nei vari campi; la linea tratteggiata rappresenta un equilibrio metastabile fra fase vapore e fase liquida soprafusa. Per la struttura e le forme polimorfiche del ghiaccio ➔ ghiaccio. La molecola dell’a., H2O, ha una struttura a forma di V (fig. 3), con una disposizione degli atomi del tipo
(con diversi sistemi si è anche potuto misurare l’angolo formato dai due legami OH che è di circa 105°).
Le proprietà chimiche e fisiche dell’a. liquida sono fortemente influenzate dalla presenza di un particolare tipo di legame nella molecola, detto legame idrogeno, che si stabilisce fra l’ossigeno di una molecola e l’idrogeno di un’altra, comportando un’aggregazione che giustifica, per es., come l’acido solfidrico, molecolarmente assai simile all’acqua (si ricordi la somiglianza fra ossigeno e zolfo presenti nello stesso gruppo del sistema periodico), presenti proprietà molto differenti. L’elevato valore della costante dielettrica dell’a. ne fa uno dei liquidi di più alto potere dissociante o ionizzante; gli acidi, le basi e molti sali in essa disciolti si dissociano, più o meno, in ioni (che vi si conservano in virtù appunto dell’alto valore della costante dielettrica e che sono della massima importanza per l’effettuarsi delle reazioni in soluzione, per i fenomeni e i processi d’elettrolisi). La stessa a. è dissociata, debolmente, nei suoi ioni e questo spiega i fenomeni d’idrolisi che avvengono in essa. In fase vapore la molecola H2O subisce un’altra dissociazione, termica, secondo lo schema
H2O ⇄ H2 + ½ O2,
che si manifesta però a temperature superiori ai 2000 °C. L’a. ha proprietà solventi verso la maggior parte dei composti; gli acidi, gli ossidi, i sali anidri si sciolgono con sviluppo di calore (causa l’idratarsi dei loro ioni), mentre i sali idrati in genere si sciolgono producendo un raffreddamento della soluzione. Dalle soluzioni acquose molti sali cristallizzano anidri mentre altri cristallizzano trattenendo a. in un dato rapporto, formando cioè dei sali idrati definiti. L’a. si combina con molti metalli (con quelli alcalini a freddo, con altri a caldo), con gli ossidi (formando gli idrossidi), con le anidridi (formando gli acidi).
Si definisce a. di costituzione quella che si combina chimicamente con un composto (per es. con un ossido metallico o con un’anidride per formare rispettivamente un idrossido o un acido) e che viene eliminata solo a temperatura piuttosto elevata; l’ a. di cristallizzazione è quella presente in un composto chimico, di solito detto idrato; viene addizionata al composto, inorganico od organico; tenuta legata da legami più deboli è presente in proporzioni diverse ma definite; si elimina di solito al di sotto di 100 °C, solitamente con un’alterazione della forma cristallina del composto; l’ a. d’impregnamento (o di assorbimento) è contenuta in determinati minerali, a seguito di un lento impregnamento, all’atto dell’estrazione dal giacimento (per es. in talune bauxiti più del 10%); l’ a. d’interposizione è quella che rimane racchiusa o interposta fra i cristalli di un composto durante la sua cristallizzazione: non ha quindi rapporti di composizione chimica con il composto e quando questo viene riscaldato l’eliminazione dell’acqua dà origine a crepitazioni.
A. naturali
La composizione chimica delle a. naturali dipende dalla natura dei terreni da cui sgorgano o vengono raccolte; fra i numerosissimi elementi chimici che possono essere presenti, i principali sono: cloro, bromo, carbonio, fosforo, azoto, boro, arsenico, sodio, potassio, litio, calcio, stronzio, bario, magnesio, ferro, manganese, alluminio, silicio, zolfo, iodio, argento. Le a. naturali contenenti solfato di calcio (selenite) sono dette selenitose.
Caratteristiche chimico-fisiche
Le principali caratteristiche chimiche e fisiche delle a. naturali, dalle quali dipende poi il loro impiego, sono: acidità, aggressività, durezza, odore, residuo fisso o salinità, sapore, temperatura, torbidità. L’acidità è dovuta alla presenza di acidi disciolti (acido carbonico, acidi umici, acido solforoso, solforico, borico, solfidrico, cloridrico, ecc.). Essa viene espressa in pH (➔). Le a. si distinguono in acide (pH < 7), neutre (pH = 7), alcaline (pH > 7). L’aggressività dipende dalla presenza dell’anidride carbonica e dei sali di calcio, poiché sussiste un equilibrio del tipo
Ca(HCO3)2 ⇄ CaCO3 + CO2 + H2O
fra bicarbonato e carbonato di calcio. Se l’acqua contiene meno anidride carbonica di quella che compete all’equilibrio precedente, parte del bicarbonato presente tende a decomporsi liberando anidride carbonica, facendo precipitare carbonato di calcio ( a. incrostante); se la quantità di anidride carbonica è superiore a quella dell’equilibrio, questa tenderà a sciogliere carbonato di calcio ( a. aggressiva). La durezza è una caratteristica conferita dalla presenza di sali di calcio e di magnesio (solfati, bicarbonati e cloruri). Nel fissare le unità di misura della durezza di un’acqua si è stabilito di riferirsi a standard convenzionali. I gradi francesi (°F) esprimono i grammi di carbonato di calcio virtualmente presenti in 100 litri di a.; i gradi tedeschi (°T) esprimono i grammi di ossido di calcio virtualmente presenti in 100 litri di a.; i gradi inglesi (°I) esprimono i grammi di CaCO3 virtualmente presenti in 70 litri di acqua. La conversione tra le diverse unità di misura si ottiene per mezzo delle relazioni: 1 °F = 0,56 °T = 0,7 °I. Si suole distinguere una durezza totale, data dalla totalità dei sali di calcio e di magnesio contenuti nell’a. a temperatura normale, una durezza permanente, data dalla percentuale di sali di calcio e di magnesio rimasti in soluzione dopo l’ebollizione (in gran parte solfati), e una durezza temporanea data dalla differenza fra durezza totale e permanente, rappresentata di solito da carbonati. In base alla durezza le a. si distinguono in dolci o molli (da 0° a 14 °F), dure (da 14° a 54 °F) e durissime (più di 54 °F). La durezza è uno dei più importanti caratteri per giudicare il possibile impiego delle a.: l’a. dura, per es., è consigliabile in conceria ma è inadatta all’impiego nelle lavanderie e nell’industria tessile; inoltre può essere di difficile digestione come bevanda ed è poco adatta per la cucina, specie per la cottura dei legumi, perché i sali di calcio e di magnesio si combinano con la legumina (albumina dei legumi) per formare un composto durissimo all’azione dei succhi gastrici. L’odore deriva dai gas disciolti, soprattutto dall’idrogeno solforato percepibile anche in dosi molto piccole. La presenza di H2S può dipendere da gas endogeno o da sostanze in putrefazione. Il residuo fisso o salinità è la quantità delle sostanze presenti in un litro d’a.; si ottiene evaporando l’a. e seccando il residuo per quattro ore a 110 °C o a 180 °C. Il sapore, secondo le sostanze disciolte, può essere dolce, salato, acido, amaro. La temperatura dipende dalle condizioni termiche del suolo e dell’aria con cui le a. sono state a contatto; alcune a. di profondità hanno una temperatura molto più elevata della media normale e sono dette a. termali. L’a. potabile dovrebbe avere una temperatura non inferiore a 6 °C e non superiore a 14 °C. La torbidità è prodotta di solito da sostanze argillose colloidali o da idrossidi di ferro in sospensione.
Classificazione
In relazione all’ambiente in cui si trovano, le a. naturali si classificano in meteoriche e litosferiche. Le a. meteoriche traggono origine dalla condensazione del vapor acqueo atmosferico dovuto alla evaporazione delle a. superficiali; contengono una percentuale variabile di gas, di ammoniaca, libera o combinata, di nitriti, nitrati, cloruri ecc.; inoltre contengono in sospensione pulviscolo atmosferico (➔ precipitazione). Le a. litosferiche sono sia superficiali sia sotterranee.
Le a. superficiali, secondo il principio che comunemente si adotta, si distinguono in a. di fiume, mare, lago e sorgente. Le a. di fiume sono correnti costituite da a. sorgive naturali miste a quelle provenienti dallo scioglimento delle nevi e dei ghiacciai e alle a. di pioggia; la composizione chimica è molto variabile: in generale contengono da 0,15 a 0,3 g per litro di sostanze disciolte e quantità molto variabili di detriti inorganici e organici. L’a. di mare è una soluzione salina diluita contenente un gran numero di elettroliti con piccole quantità di materie azotate e minime percentuali di colloidi; un problema che solleva grande interesse è quello della dissalazione dell’a. di mare, sia a usi potabili sia a usi industriali e agricoli. Le a. dei laghi salati hanno proprietà simili a quelle dei mari di cui possono considerarsi una segmentazione; le a. dei laghi dolci hanno proprietà corrispondenti a quelle dei fiumi che vi affluiscono. Le a. di sorgenti (o minerali) sono a. sotterranee che affluiscono naturalmente all’esterno; a seconda della natura delle rocce con cui sono state a contatto nel sottosuolo, della loro origine e della profondità da cui provengono possono risultare anche molto diverse per composizione chimica e per temperatura (a. fredde se di temperatura inferiore a 20 °C, calde o termali se la loro temperatura supera 20 °C). A seconda di determinate caratteristiche, prese in considerazione soprattutto dalla geografia fisica, le a. superficiali possono poi distinguersi in incanalate e dilavanti (dette anche selvagge) a seconda che scorrano o no sulla superficie terrestre in un solco o canale di raccolta, o comunque in una direzione ben definita. La loro azione è varia e incostante e il risultato finale è il dilavamento della superficie del terreno che si manifesta con l’asportazione dal suolo dei materiali mobili minuti o grossolani e si effettua sia sui terreni poco inclinati, sia su tutto il versante di un monte: è assai debole se la pendenza del terreno è minima o massima; si accresce invece con le pendenze medie. Si viene comunque a originare una complessa rete di venule acquee che confluiscono verso un solco ben definito, quello torrentizio.
Le a. sotterranee provengono dalle a. meteoriche che penetrano nel sottosuolo sollecitate dalla gravità, dalla condensazione nel sottosuolo del vapor d’a. contenuto nell’aria che circola negli strati superiori della litosfera (zona di aerificazione) e dalle a. endogene di nuova formazione che si originano dalla combinazione di idrogeno con ossigeno ad alta temperatura. La nomenclatura delle a. sotterranee non è molto uniforme; restando nei criteri di classificazione più in uso possono distinguersi in:
a) a. del suolo vegetale, sotto qualsiasi forma;
b) a. vadose, a. meteoriche che scendono nel sottosuolo per azione della gravità;
c) a. di cava, a. della zona di aerificazione non soggette a gravità, che occupano minuti interstizi delle rocce: distinte in pellicolari se aderiscono alla superficie di pori e di piccole fessure delle rocce, e capillari se riempiono fessure e interstizi capillari; tali a. sono dette di cava perché visibili durante i lavori di estrazione di materiali e rocce;
d) a. freatiche, a. libere, correnti o stagnanti, non soggette a pressione perché contenute in strati permeabili limitati inferiormente da terreno impermeabile; per essere captate devono quindi essere direttamente raggiunte; tuttavia possono spontaneamente sgorgare all’esterno se lo strato acquifero (detto falda) viene a essere interrotto dalla superficie topografica del terreno; il livello di tali a. è variabile con il variare delle stagioni;
e) a. artesiane, a. libere, correnti o stagnanti, racchiuse fra due strati inclinati di terreno impermeabili, portate in pressione in dipendenza del carico; esse possono quindi raggiungere la superficie esterna quando si perfori lo strato impermeabile superiore;
f) a. salienti, a. comprese in terreni permeabili, che vengono a trovarsi tra due strati impermeabili ondulati o con curvature rivolte verso l’alto e quindi sottoposte a un ‘carico piezometrico’ il quale le sospinge come in una condotta forzata: se perciò nel suolo si hanno fessurazioni o pozzi artificiali in un settore a ‘carico’ sensibile, le a. risalgono in superficie e talvolta zampillano anche sino a una certa altezza;
g) a. fossili, a. incluse nelle rocce (a. salata dei giacimenti petroliferi) e anche nei minerali (per es. nei cristalli di quarzo) dall’epoca della loro formazione;
h) a. giovanili, precedentemente definite, che alcuni distinguono in a. plutoniche, se si separano da un magma profondo in via di consolidazione, e a. vulcaniche, se si sprigionano dalla lava in via di consolidazione.
Le a. sotterranee vengono anche qualificate in base a quelle caratteristiche considerate importanti dalla geografia fisica.
A. ipogeiche
A. meteoriche che attraverso le permeazioni del suolo passano in profondità, aumentando la loro temperatura, specie in prossimità di focolai vulcanici profondi, e mineralizzandosi, e poi risalgono, attraverso altre fessure, rapidamente in superficie; appartengono al gruppo delle a. termali ma non presentano di solito temperature esterne superiori a 50 °C; sono ricche di gas e di sali minerali disciolti, che non trovano riscontro nelle rocce superficiali dalle quali emergono.
A. profonde
A. che si accumulano nelle parti basali dei massicci calcarei e, per i fenomeni di fratturazione in grande delle masse e per quelli di solubilizzazione, possono dare luogo a canalizzazioni notevoli, complesse, a livelli differenti, nelle quali corrono con regime incostante e variabilissimo a causa degli ostacoli che si presentano al loro libero decorso.
A. di base
Ipotetica falda acquea che, al livello medio marino, si estenderebbe negli strati rocciosi e che dovrebbe perciò trovarsi in ogni terreno benché a profondità diverse per effetto della capillarità: la sua esistenza è ammessa da alcuni autori e negata da altri.
A. potabile
Per poter essere destinata agli usi alimentari e domestici un’a. deve rispondere a determinati requisiti di purezza oltre che a quelli organolettici, chimici e fisici: non deve contenere affatto o quasi batteri ed essere comunque priva di germi patogeni. Pertanto per giudicare della potabilità di un’a. occorre sottoporla ad analisi fisica, chimica, batteriologica, organolettica, e spesso procedere anche allo studio della natura e delle caratteristiche del terreno ove l’a. scorre. Quasi il 90% dell’a. consumata in Italia per uso potabile (circa 280 litri per abitante e per giorno) proviene da sistemi idrici sotterranei. Entro il 2025 è previsto in Italia un raddoppio del consumo, con una significativa crescita percentuale del prelievo di a. superficiali.

Poiché non sempre le a. a disposizione per uso potabile (specie quelle superficiali) presentano tutti i necessari requisiti richiesti, prima di essere immesse nell’uso esse devono venire sottoposte a speciali trattamenti che ne migliorino le caratteristiche chimiche, fisiche, organolettiche. Tali trattamenti, che si effettuano generalmente in impianti centrali, comprendono operazioni di chiarificazione, filtrazione, sterilizzazione e, più raramente, operazioni tendenti a eliminare l’eccessiva quantità di sali di ferro, di calcio e di magnesio o i cattivi odori. La chiarificazione viene eseguita lasciando riposare l’a. in grandi bacini di sedimentazione; per accelerare la separazione delle sostanze in sospensione si possono aggiungere sostanze coagulanti e flocculanti (solfato di alluminio, cloruro ferrico ecc.) che destabilizzano le particelle sospese (neutralizzandone la carica elettrica superficiale) e danno luogo a idrossido di alluminio o di ferro ecc., sotto forma di flocculi che si separano piuttosto rapidamente trascinando le impurezze sospese. Per migliorare l’efficienza del trattamento di coagulazione-flocculazione si aggiungono, talvolta, sostanze coadiuvanti (silice attivata, polielettroliti) che consentono di ottenere flocculi sedimentabili con grande rapidità. Il tempo complessivo richiesto dalla coagulazione-flocculazione e dalla sedimentazione è notevolmente abbreviato facendo avvenire queste operazioni in una apparecchiatura, detta decantatore-acceleratore (fig. 4), in cui i flocculi si accrescono in presenza di sostanze già precipitate.
La filtrazione si effettua mediante passaggio dell’a. attraverso letti di granuli di sabbia o di carbone nei quali si depositano, durante il deflusso dell’a., le sostanze in sospensione. Queste formano una massa melmosa che facilita l’arresto delle impurezze batteriche (fino al 99%). La distruzione dei germi patogeni si può ottenere con mezzi chimici o fisici. Nel primo caso si aggiungono all’a. sostanze (ozono, cloro gassoso o composti clorurati come gli ipocloriti, le clorammine, ecc.) dotate di azione ossidante, capaci cioè di dar luogo a sviluppo di ossigeno che distrugge le sostanze organiche presenti e quindi anche gli eventuali microbi. L’uso di ipoclorito di sodio o di calcio se in eccesso, anche piccolo, può dare all’a. sapore e odore sgradevole: si provvede allora alla declorazione dell’a. mediante filtrazione su carboni attivi adsorbenti. L’impiego delle clorammine ha il vantaggio di combattere la formazione di odori e sapori sgradevoli e produrre un’azione sterilizzante più lenta ma persistente. Per sterilizzare un’a. si può ricorrere all’ebollizione che però elimina i gas disciolti e fa intorbidare l’a. per la precipitazione di carbonato di calcio con conseguente alterazione del suo sapore e della sua digeribilità. Si può anche sfruttare l’azione microbicida delle radiazioni ultraviolette facendo passare l’a. da depurare davanti a lampade a vapori di mercurio (che emanano una luce ricca di radiazioni ultraviolette) o immergendo queste lampade nell’acqua. Per eliminare odori, colori e sapori sgradevoli, talvolta si filtra l’a. su un letto di carbone attivo dotato di elevate proprietà adsorbenti. L’eliminazione dei sali di ferro, di calcio e magnesio è richiesta raramente per le a. destinate a usi potabili, per il relativamente basso tenore presente; essa si pratica invece frequentemente per le a. usate dalle industrie.
Il trattamento delle a. destinate a uso potabile sta diventando sempre più complesso a causa della presenza di microinquinanti organici provenienti da fonti sia diffuse, sia puntuali. Le fonti diffuse riguardano le attività agricole e in particolare l’uso di antiparassitari. Le fonti puntuali sono associate a scarichi di insediamenti civili e produttivi, a discariche non controllate, ad aree industriali dismesse ecc., con conseguente presenza nelle a. di composti organici (per es., idrocarburi clorurati alifatici a basso peso molecolare, quali tetracloroetilene, tricloroetilene, 1,1,1-tricloroetano, largamente usati come solventi, nella preparazione di fitofarmaci ecc.). Un’altra fonte di contaminazione delle a. potabili è collegabile alla formazione di composti secondari indesiderati dovuti agli stessi processi di potabilizzazione; in particolare, la disinfezione con cloro o con ipoclorito dà luogo, in presenza di precursori organici (soprattutto substrati organici naturali sempre presenti nelle a. superficiali), a cloroformio e ad altri trialometani; differenti tipi di disinfettanti (biossido di cloro, ozono, a. ossigenata, radiazione UV, acido peracetico), pur producendo minori quantità di composti indesiderati, non sono esenti da inconvenienti
A. minerali
Non risulta facile dare una definizione univoca di a. minerale, anche perché ne esistono numerosissime, con i più diversi caratteri chimici e fisici. La normativa italiana (d.m. 1° febbraio 1983) stabilisce una classificazione delle a. minerali in base alla quale alla denominazione «acqua minerale naturale» può essere aggiunta una delle seguenti dizioni: «oligominerale» o «leggermente mineralizzata», quando il contenuto salino, calcolato come residuo fisso a 100 °C, non è superiore a 500 mg/l; «minimamente mineralizzata», per valori inferiori a 50 mg/l; «ricca di sali minerali», per valori superiori a 1500 mg/l. In relazione poi al contenuto dei singoli ioni le a. minerali possono essere ulteriormente distinte in: a) «contenente bicarbonato», se il tenore di bicarbonato è superiore a 600 mg/l;
b) «solfata», se il tenore di solfati è superiore a 200 mg/l;
c) «clorurata», se il tenore di cloruro è superiore a 200 mg/l;
d) «calcica», se il tenore di calcio è superiore a 150 mg/l;
e) «magnesiaca», se il tenore di magnesio è superiore a 50 mg/l;
f) «fluorurata» o «contenente fluoro», se il tenore di fluoro è superiore a 1 mg/l;
g) «ferruginosa» o «contenente ferro», se il ferro bivalente è superiore a 1 mg/l;
h) «sodica», se il tenore di sodio è superiore a 200 mg/l;
i) «indicata per le diete povere di sodio», se il tenore di sodio è inferiore a 20 mg/l.
Nella pratica commerciale e domestica si usa comunque fare una distinzione tra a. minerali medicamentose e a. minerali da tavola. Le prime, caratterizzate da particolari proprietà terapeutiche derivanti dalla loro composizione chimica, hanno un uso limitato a casi specifici, mentre le seconde sono quelle di maggior consumo, in quanto utilizzate come bevande abituali in sostituzione della comune a. potabile
A. distillata
Acqua chimicamente pura ottenuta mediante la depurazione di quella comune; più corretta, ma meno usuale, è la denominazione a. deionizzata o a. demineralizzata. La depurazione si può realizzare per evaporazione (impropriamente detta distillazione) o con altri sistemi (scambio ionico, elettrodialisi, osmosi inversa). L’evaporazione di piccole quantità di a. si compie in caldaie di rame; in laboratorio si ricorre ad apparecchi di vetro o di vetro di quarzo o anche (per usi speciali) di rame internamente argentato. Generalmente non si utilizzano le prime porzioni dell’a. che evapora e si arresta il procedimento quando si è evaporato il 75-80% dell’a. di partenza; per distruggere le eventuali sostanze organiche si aggiunge un po’ di permanganato potassico ecc. Per l’evaporazione di notevoli quantitativi di a. (per industrie farmaceutiche, alimentari, alimentazione di caldaie ad alta pressione, elettrolizzatori, accumulatori ecc.) si ricorre a evaporatori a multiplo effetto o a termocompressione. A. completamente o quasi demineralizzata si può avere anche con resine scambiatrici.
Agraria
L’irrigazione delle terre coltivate costituisce in tutto il mondo circa il 75% del prelievo idrico totale, mentre le a. prelevate per l’industria incidono per circa il 20% e le a. per uso civile per circa il 5%. In Italia l’a. per l’irrigazione proviene per il 60-70% dai fiumi e per la parte restante da pozzi, fontanili e serbatoi d’invaso; il consumo idrico irriguo è concentrato nei mesi primaverili-estivi. Una notevole percentuale di a. (circa il 55-65%) torna all’atmosfera attraverso i processi di evaporazione (dalle superfici idriche e dal terreno) e di traspirazione (dalla copertura vegetale); un’altra parte torna disponibile ( a. di ritorno), in quanto va a ricaricare le falde sotterranee o affluisce negli alvei vallivi dei corsi d’acqua. Il consumo irreversibile di a. per l’irrigazione è pari a 2600-2700 m3t−1 di prodotto agricolo (valore medio mondiale calcolato tenendo conto di tutte le colture); esso è destinato a diminuire in misura significativa grazie al miglioramento delle tecniche irrigue (nel lungo periodo si prevede possa scendere fino a 1000 m3t−1).
Biologia
L’a. è il costituente inorganico più abbondante degli organismi viventi. I valori minimi del contenuto idrico si riscontrano negli insetti (10%), mentre per gli animali terrestri sono dell’ordine del 60-70%. Per l’uomo adulto diversi autori danno valori variabili fra il 58 e il 67%, con una notevole diversità nei vari organi, tessuti e liquidi. In via del tutto schematica, nell’a. di un organismo si è soliti distinguere due frazioni: l’ a. libera e l’ a. legata o fissa. La prima è il più cospicuo componente dei liquidi organici (sangue, linfa, liquor, succhi interstiziali, succhi intercellulari, secreti), la seconda fa parte integrante dei tessuti e dei liquidi dell’organismo sia come a. di imbibizione dei biocolloidi sia come a. di idratazione di numerosissimi altri biocostituenti organici e inorganici, sia infine entrando nella composizione di alcune molecole organiche in rapporti definiti e costanti. Da un punto di vista chimico-fisico il plasma cellulare in stato di attività può considerarsi un sistema eterogeneo di tipo colloidale comprendente una fase dispersa (essenzialmente proteica) e una fase acquosa disperdente.
All’a. e all’interazione fra questa e qualunque altro biocostituente sia inorganico sia organico spetta un ruolo assolutamente essenziale per una qualsiasi attività di un qualsiasi sistema biologico. Tale ruolo deriva dalla struttura e dalle proprietà chimico-fisiche dell’a.: essa è particolarmente adatta come solvente di composti inorganici e organici negli organismi viventi; avendo una costante dielettrica elevata, permette la dissociazione degli elettroliti; avendo una capacità termica elevata, può funzionare da liquido termostatico; il suo alto calore latente di evaporazione è responsabile della sua capacità termoregolatrice. Per queste ragioni è facile capire come il metabolismo cellulare (e le funzioni da esso derivanti) sia strettamente connesso con il contenuto idrico degli organismi. Da un punto di vista più strettamente biochimico, l’essenziale importanza dell’a. deriva dalla considerazione che condizione generale per lo svolgimento delle trasformazioni metaboliche è che le sostanze reagenti siano allo stato di soluzione acquosa. L’a. inoltre costituisce un vero e proprio reagente in moltissime reazioni del chimismo cellulare: per es. l’a. viene utilizzata nei numerosi processi di scissione enzimatica di legami che avvengono durante l’utilizzazione dei carboidrati, dei lipidi, delle proteine e negli svariati processi di degradazione. A. è continuamente generata ogni qualvolta la cellula utilizza l’ossigeno quale accettore finale di elettroni nei processi di ossidazione biologica.
L’importanza della differenziazione tra a. non legata, adsorbita, fisicamente trattenuta e legata chimicamente, ottenibile con metodi di analisi strumentale (RMN e termoanalitici), emerge chiaramente in campo alimentare. Ai fini della conservazione di un alimento infatti l’eliminazione dell’a. libera, ossia non impegnata in equilibri chimico-fisici a livello cellulare, quanto più è completa tanto più è utile per inibire le attività chimiche e microbiologiche; l’a. combinata o fissata non deve invece essere eliminata, affinché gli equilibri fra acqua e tessuti non vengano compromessi; le variazioni del suo contenuto allo stato libero e combinato sono importanti per il mantenimento delle proprietà organolettiche degli alimenti.
Diritto
A. pubbliche
I laghi, i fiumi, i torrenti e le altre a. definite pubbliche dalle leggi in materia fanno parte nel demanio idrico, previsto espressamente all’articolo 822 del codice civile. Più in particolare, rientrano della categoria del «demanio naturale necessario» e appartengono quindi allo Stato oppure, in casi eccezionali, alle Regioni a statuto differenziato, o alle Regioni a statuto ordinario nel caso di attribuzione dei porti lacuali e di navigazione interna. Sono altresì definiti beni pubblici a fruizione collettiva, nel senso che appartengono, sulla base di una riserva originaria di legge, all’ente pubblico territoriale che si occupa della loro gestione e conservazione ma sono destinati all’uso da parte della collettività in generale.
Il Testo Unico delle a. pubbliche approvato con r.d. 1775/1933, stabilisce che sono pubbliche tutte le a. sorgenti, fluenti e lacuali, anche se artificialmente estratte dal sottosuolo, sistemate o incrementate, le quali, considerate sia isolatamente per la loro portata o per l’ampiezza del rispettivo bacino imbrifero, sia in relazione al sistema idrografico al quale appartengono, abbiano o acquistino attitudine a usi di pubblico, generale interesse. La l. 36/1994 (cosiddetta legge Galli) relativa alle disposizioni in materia di risorse idriche ha ampliato la categoria delle a. pubbliche includendovi tutte le a. superficiali e sotterranee ancorché non estratte dal sottosuolo (art. 1). In tal senso il d.p.r. 238/1999 afferma l’appartenenza allo Stato di tutte le a., sotterranee e superficiali, anche raccolte in vasi e cisterne, escluse quelle piovane non convogliate in un corso d’a. o non ancora raccolte in vasi o cisterne.
La gestione delle a. pubbliche avviene in base a una ripartizione di competenze tra Stato, regioni ed enti locali. In proposito il d. legisl. 112/1998, attribuisce alle regioni e agli enti locali una competenza generale sulla gestione del demanio idrico con conferimento di tutte le funzioni non espressamente riservate allo Stato. Tra queste, la progettazione, realizzazione e gestione di opere idrauliche di qualsiasi natura; le funzioni di polizia idraulica e di pronto intervento; la programmazione, pianificazione e gestione integrata degli interventi di difesa delle coste e degli abitati costieri; le funzioni relative alle concessioni di estrazione di materiale litoide dai corsi d’a., di spiagge lacuali, di pertinenze idrauliche e di aree fluviali. Tale decreto conferisce espressamente allo Stato le funzioni che necessitano di una gestione a livello nazionale, tra cui: il censimento nazionale dei corpi idrici; la programmazione e il finanziamento degli interventi di difesa del suolo; la programmazione della razionale utilizzazione delle risorse idriche, dei trasferimenti di a. e degli usi plurimi delle a.; l’individuazione delle aree a rischio di crisi idrica, con finalità di prevenzione delle emergenze idriche; la determinazione dei criteri per la gestione del servizio idrico integrato; la formazione del bilancio idrico nazionale. Il diritto di utilizzazione delle a. pubbliche per obbiettivi connessi alla produzione agricola e industriale può essere attribuito, sia a enti pubblici sia a soggetti privati, attraverso una concessione amministrativa. Le controversie in materia di a. pubbliche sono devolute alla giurisdizione dei tribunali delle acque.
Negli ultimi anni si è manifestata una progressiva riduzione delle risorse idriche e si è evidenziata, a livello comunitario e internazionale, la necessità di una maggiore salvaguardia e tutela del bene-a., di uno sfruttamento razionale di esso nell’ambito di un progetto di gestione globale, da un punto di vista quantitativo e qualitativo. In linea con tale tendenza, la legge Galli, ha provveduto all’ampliamento della categoria delle acque pubbliche e ha affermato che l’utilizzazione delle stesse deve essere effettuata secondo criteri di solidarietà in modo da tutelare le aspettative e i diritti delle generazioni future. A tal fine gli usi delle a. devono essere indirizzati al risparmio e al rinnovo delle risorse, per non pregiudicare il patrimonio idrico, la vivibilità dell’ambiente, l’agricoltura, la fauna e la flora acquatiche, i processi geomorfologici e gli equilibri idrogeologici. In particolare, l’uso dell’a. per il consumo umano è prioritario rispetto agli altri e qualsiasi ulteriore utilizzazione è subordinata alla soddisfazione di tale bisogno e alla non alterazione della qualità dell’acqua.
Religione
L’a. è universalmente oggetto dell’interesse dell’uomo religioso sia sul piano delle idee e delle immagini, nell’ambito del quale dà origine a una serie di innumerevoli figure e racconti mitici, sia su quello cerimoniale e rituale in cui trova un larghissimo impiego. Su tutti e due i piani e presso tutte le civiltà religiose si riscontra una tendenza univoca a scorgere e riconoscere tradizionalmente nell’a. caratteristiche e proprietà affini e ben determinate.
Ovunque l’a. è vista come segno del virtuale e del preformale e al tempo stesso come origine e sorgente di tutte le possibili forme. Ma è nella pratica religiosa, nei riti e nelle cerimonie che l’a. rivela in maniera saliente la sua capacità di restare riserva inesauribile per nuove creazioni e nuove metamorfosi. Nei riti di immersione in tutte le loro forme la valenza simbolica dell’a. è sentita capace di sottrarre l’individuo dalla sua condizione profana per riportarlo all’incontaminata indifferenziazione delle origini: di lì è possibile ricominciare una nuova esistenza, secondo un’idea centrale che sta alla base di tutte le pratiche e le concezioni catartiche che comprendono l’uso dell’acqua. In una dimensione diversa da questa prospettiva, generalmente a carattere femminile, preformale, germinativo si collocano le nozioni, i racconti mitici e le esperienze religiose relativi alla pioggia, che viene rappresentata per lo più come principio fecondatore attivo, d’origine celeste e maschile.
Tecnica
A. per usi industriali
Caratteristiche e utilizzo
Gli impieghi dell’a. nell’industria sono molteplici, così come diverse sono le caratteristiche richieste per ciascun utilizzo. Solitamente si distingue tra a. di processo, a. di raffreddamento e a. per vapore. I requisiti per un’ a. di processo sono direttamente legati al tipo di industria e variano in maniera notevole (si va dall’a. distillata nell’industria farmaceutica all’a. potabile nell’industria alimentare o alla comune a. superficiale nell’industria metallurgica); per quanto riguarda invece l’ a. di raffreddamento, in molti casi può essere impiegata anche quella di mare, essendo la bassa temperatura il suo requisito fondamentale; infine l’ a. per vapore deve essere necessariamente priva di sali per evitare fenomeni di incrostazione e di corrosione negli impianti.
Le attività industriali con i maggiori consumi di a. sono: l’industria siderurgica (30-35%), l’industria chimica (20-25%), l’industria cartaria (circa il 15%), l’industria petrolifera (circa il 10%). Negli stabilimenti industriali (con consumo di a. superiore a 0,5 m3/s) i fabbisogni idrici si ripartiscono orientativamente nel modo seguente: 36% per il processo, 26% per i servizi e gli usi sanitari, 23% per il raffreddamento, 12% per le caldaie, 3% per altri usi. Questi dati di consumo hanno valore soltanto orientativo in quanto dipendono in misura rilevante dagli accorgimenti tecnologici adottati nel processo produttivo. Al riguardo, particolarmente efficace risulta il ricircolo di una parte dell’a. già utilizzata; in Italia, il grado di ricircolo, definito come il rapporto fra l’a. impiegata e l’a. prelevata, è attualmente prossimo a 2, ma appare suscettibile di essere aumentato.
Per la sua disponibilità in natura e per l’elevato calore specifico l’a. è quasi sempre preferita a ogni altra sostanza come fluido di raffreddamento nelle apparecchiature di scambio termico a contatto indiretto: tale impiego non inquina l’a. avendo l’unico effetto di aumentarne la temperatura. Pertanto, anche per contenere in misura ragionevole i consumi specifici di a. per l’industria, l’a. di raffreddamento è quasi sempre riutilizzata dopo un trattamento che ne diminuisce la temperatura; esso si realizza in torri dove l’a., venendo a contatto con aria, evapora parzialmente e così si raffredda. A causa dell’esposizione all’aria e dell’effetto di concentrazione dovuto alla conseguente evaporazione, l’a. può divenire incrostante e, soprattutto, corrosiva; conviene pertanto aggiungere qualche inibitore di corrosione (cromati, polifosfati ecc.).
Impianti termici
Per l’elevato calore specifico, che le consente d’immagazzinare energia termica, per la debole viscosità che ne facilita la circolazione entro tubazioni e per il debole potere corrosivo riguardo a queste, l’a. si presta come mezzo di scambio termico per impianti di riscaldamento (ad a. o a vapore acqueo) e come fluido evoluente nelle macchine termiche a vapore, fisse o mobili, alternative (motrici a vapore) o rotative (turbine a vapore). L’a. destinata a tali impianti deve contenere la minore quantità possibile di sali disciolti, specie di calcio, magnesio, silicio ecc. Questi infatti producono incrostazioni che, essendo cattive conduttrici del calore, determinano maggior consumo di combustibile e, se di spessore considerevole, possono provocare arroventamento delle lamiere (con conseguente facile logorio) e pericolo di esplosioni quando, per la rottura dell’incrostazione, l’a. viene improvvisamente a contatto del metallo surriscaldato. Al fine di evitare la formazione di incrostazioni, si aggiungono all’a. già depurata correttivi chimici.
Impianti idroelettrici e idraulici
L’energia cinetica acquistata nella caduta da masse d’a. opportunamente convogliate è utilizzata nelle centrali idroelettriche per la produzione di energia elettrica. Utilizzazione di energia posseduta dall’a. si ha anche nei vari tipi di motori idraulici. L’a. venne adoperata in passato anche in impianti di trasporto (funicolari, ascensori idraulici, ecc.) per costituire un contrappeso atto ad assicurare un regolare funzionamento dell’impianto. Ovvi inconvenienti di sicurezza, di rapidità, d’ingombro hanno suggerito l’abbandono di tali sistemi per altri più moderni; così come l’impiego dell’a. per la trasmissione di pressioni a distanza (per es., accumulatore idraulico) va cadendo in disuso a favore di liquidi più adatti (oli e miscele incongelabili meglio atte a evitare corrosioni, incrostazioni e fughe).
A. di rifiuto
Caratteristiche e generalità
Sono le a. provenienti dallo scarico di abitazioni, officine, industrie ecc., e che contengono pertanto sostanze estranee. Sono dette a. di scarico, a. reflue, o anche liquami, effluenti idrici. Le a. dei centri abitati si dicono nere o luride (quelle contenenti deiezioni umane) e bianche (quelle meteoriche ecc.) e possono essere incanalate in un’unica o in due distinte fognature. Le a. di rifiuto industriali si possono distinguere secondo che contengano prevalentemente sostanze organiche più o meno facilmente putrescibili o principalmente sostanze inorganiche (sali, acidi, basi). Con il crescere della popolazione nei centri urbani e con l’aumentare delle industrie, lo smaltimento delle a. di rifiuto ha posto notevoli problemi igienico-sanitari, in quanto dette a., convogliate dalle fogne, non possono immettersi liberamente nei corsi d’a. perché ne danneggiano la vita acquatica e ne pregiudicano le possibili utilizzazioni (attività ricreative, irrigazione, ecc.): pertanto richiedono opportuni trattamenti di depurazione.
Depurazione
Le a. di rifiuto di origine domestica sono dette fresche se contengono ancora ossigeno disciolto sia pure in quantità minima, settiche se in esse sono già presenti processi di decomposizione anaerobica. La quantità di ossigeno richiesta dai processi biochimici di depurazione (BOD, Biochemical Oxygen Demand) è di 180-540 ppm, contengono 200-600 ppm di sostanze sospese e 30-90 ppm di azoto totale; tali valori corrispondono a consumi di a. pro capite compresi fra 100 e 300 l al giorno (ovviamente all’aumentare del consumo diminuiscono le concentrazioni). La depurazione delle a. di rifiuto di origine domestica può essere realizzata in tre stadi successivi: a) il trattamento primario, che consente di allontanare dai liquami la maggior parte delle sostanze sospese e galleggianti; b) il trattamento secondario, che consente di allontanare dai liquami la maggior parte delle sostanze organiche disciolte di tipo colloidale; c) il trattamento terziario, che consente di eliminare le sostanze non allontanate con i trattamenti precedenti (tensioattivi non biodegradabili, pesticidi, composti azotati, fosfati ecc.).
Le principali operazioni che intervengono nel trattamento primario sono: la grigliatura, che trattiene le sostanze sospese grossolane (reimmesse nelle a. di rifiuto dopo triturazione), la dissabbiatura, che rimuove i materiali silicei, la disoleazione e la sgrassatura, che allontanano le sostanze oleose e grasse, la sedimentazione, che consente di eliminare dai liquami tutte le sostanze sospese aventi velocità di caduta sufficientemente elevate (sostanze sedimentabili).
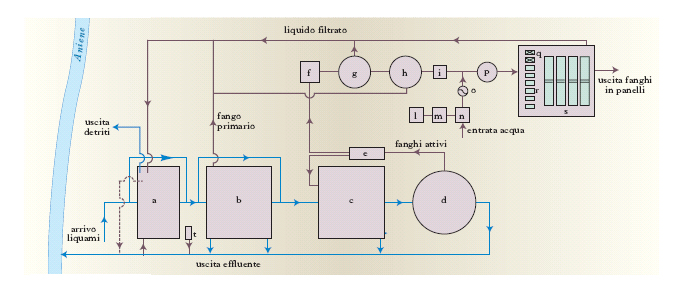
Il trattamento secondario
utilizza processi biologici di depurazione (generalmente aerobici) analoghi ai processi naturali di autodepurazione, ma realizzati in apparecchiature dove le condizioni operative sono tali da rendere molto più elevate le velocità di degradazione delle sostanze organiche a opera dei microrganismi (biomassa). Nei reattori in cui avvengono i processi depurativi biologici i microrganismi possono essere dispersi, sotto forma di biofiocchi, in seno al liquame da depurare (reattori con biomassa dispersa) o essere ancorati, in forma di film biologico, su superfici inerti (reattori a biofilm). I principali processi depurativi che utilizzano reattori con biomassa sospesa sono: l’ossidazione con il sistema a fanghi attivati, la depurazione in lagune (aerate e non), l’ossidazione con il sistema a pozzo profondo. Nel processo di ossidazione a fanghi attivati la dispersione dei fiocchi biologici (fanghi attivati) dentro il liquame e l’ossigenazione della miscela fanghi attivati/liquame sono realizzate da turbine di superficie (aerazione meccanica) o mediante insufflazione d’aria (aerazione a diffusione d’aria). La miscela aerata che esce dal reattore a fanghi attivati viene inviata in un sedimentatore secondario in cui il liquido depurato viene separato dalla biomassa, in parte ricircolata nel reattore, in parte inviata a successivi trattamenti di stabilizzazione (fig. 5). Nella depurazione a lagune (aerate e non), il liquame da trattare permane dentro bacini di grande volume, di solito ricavati mediante scavo in terra e di profondità variabile fra 0,5 e 4 m, per tempi così lunghi da consentire soddisfacenti efficienze di rimozione delle sostanze organiche senza necessità di sedimentazione finale e di ricircolo della biomassa. Nelle lagune aerate l’ossigeno viene fornito tramite turbine galleggianti o insufflazione d’aria; nelle lagune non aerate il processo depurativo può avvenire sia aerobicamente (tramite l’ossigeno proveniente dalla riaerazione naturale della superficie liquida o dallo sviluppo algale) che anaerobicamente. Nel processo di ossidazione a pozzo profondo si utilizza un pozzo verticale scavato nel terreno, profondo anche 200 m e con diametro anche maggiore di 5 m, configurato internamente come un tubo a U, dove il liquame da trattare dapprima discende fino al fondo e poi risale fino alla sommità. Lungo il percorso discendente del liquame viene insufflata aria arricchita di ossigeno; a causa dell’aumento di solubilità dell’ossigeno dovuto alle elevate pressioni che si instaurano nelle regioni inferiori del pozzo, il processo è caratterizzato da una elevata capacità depurativa.
I principali processi depurativi che utilizzano reattori a biofilm sono:
a) l’ossidazione su filtri percolatori,
b) l’ossidazione su biodischi,
c) la depurazione in letti fluidizzati.
Nell’ossidazione su filtri percolatori, il liquame viene immesso sulla sommità di un letto poroso, costituito da materiale di riempimento (coke, antracite, ciottoli ecc., e, nei filtri più moderni, materiale plastico preformato) su cui si forma un film biologico ossigenato da una corrente di aria che attraversa il filtro per tiraggio naturale; il liquame, scorrendo sul materiale di riempimento, subisce così un processo di ossidazione biologica; sul fondo del filtro un sistema di drenaggio raccoglie il liquame depurato che viene inviato in un sedimentatore secondario. Una versione modificata dei filtri percolatori è rappresentata dai biodischi, costituiti da dischi in materiale plastico che ruotano parzialmente immersi nel liquame da trattare; sulla superficie dei dischi si forma un film biologico che per effetto della rotazione è alternativamente a contatto con l’aria (durante l’emersione) e con il liquame (durante l’immersione). Nei reattori a letto fluidizzato un letto di particelle inerti viene mantenuto in condizioni di fluidizzazione da una corrente ascendente costituita dal liquame da trattare, in cui viene disciolto ossigeno commerciale prima dell’immissione nel reattore; sulle particelle si forma un film biologico di spessore controllato. Dei processi depurativi sopra descritti quelli di gran lunga più impiegati sono l’ossidazione a fanghi attivali e la depurazione in filtri percolatori. La depurazione effettuata con il processo a pozzo profondo e con i reattori a letto fluidizzato, per quanto promettente, non ha ancora trovato una definitiva applicazione in impianti in prima scala; è in corso di sperimentazione anche la possibilità di applicare il processo di digestione anaerobica, correntemente impiegato come trattamento di stabilizzazione biologica dei fanghi, per la depurazione delle acque di rifiuto di origine domestica in particolari reattori in cui si forma un letto espanso di particelle di biomassa. In impianti di ridotta dimensione, per lo più di uso domestico o destinati a piccole comunità, possono trovare impiego le cosiddette fosse settiche (➔ fossa) o le vasche Imhoff (➔ Imhoff, Karl). Nell’ambito del trattamento secondario rientra anche la disinfezione, che consente di eliminare quasi totalmente dall’a. di rifiuto i microrganismi patogeni tramite l’aggiunta di reattivi chimici (cloro, acqua ossigenata, azoto) o l’impiego di radiazioni (ultraviolette, gamma). C di particolari destinazioni dell’effluente (reimmissione in falda, usi potabili ecc.) è necessario procedere anche a un trattamento terziario in cui intervengono processi chimicofisici (coagulazione-flocculazione, precipitazione chimica, adsorbimento su carbone attivo, scambio ionico, operazioni con membrana ecc.) e biologici (nitrificazione-denitrificazione). In particolare, quando le a. trattate vengono scaricate in corpi idrici a debole ricambio (per es., i laghi) in cui possono aver luogo fenomeni di eutrofizzazione, è necessario rimuovere il fosforo (tramite precipitazione chimica con cloruro ferrico o solfato di alluminio) e l’azoto (tramite nitrificazione, cioè ossidazione biologica dell’azoto ammoniacale, seguita da denitrificazione, cioè riduzione per via biochimica dei composti dell’azoto ossidato in azoto gassoso).
Il trattamento terziario costituisce lo stadio finale della depurazione delle a. di rifiuto che comprende anche lo smaltimento dei fanghi ottenuti durante la depurazione dei liquami (➔ fango). Si procede, infine, a una depurazione biologica naturale delle a. di rifiuto di origine domestica, distribuendo l’a. da depurare su ampie superfici di terreno coltivato o arato che agisce da filtro e fissa parte delle sostanze disciolte; l’a. viene raccolta a pochi metri di profondità mediante apposita rete di tubi di drenaggio. Questo metodo naturale di depurazione trova sempre minori applicazioni per mancanza di terreni adatti, per le ampie superfici necessarie, per gli elevati costi di trasporto.
Per quanto concerne la depurazione delle a. di scarico industriali ➔ scarico.