
colloidale, stato
Stato di aggregazione caratteristico, distinto dalle soluzioni e dalle sospensioni: una sostanza è in tale stato quando è dispersa in un’altra sostanza sotto forma di particelle generalmente amorfe, di dimensioni comprese approssimativamente fra 10–5 e 10–7 cm, corrispondenti a raggruppamenti di 103-109 atomi, non visibili al microscopio (al contrario delle sostanze in sospensione), che diffondono molto lentamente attraverso membrane porose. Il termine colloide fu introdotto dal chimico inglese T. Graham per distinguere tali sostanze da quelle, come lo zucchero, che diffondono facilmente attraverso le membrane, da lui chiamate cristalloidi. Lo stato colloidale può essere considerato uno stato disperso, non perfettamente inquadrabile né tra i sistemi omogenei caratterizzati dalla presenza di una sola fase né tra quelli eterogenei a più fasi. Gran parte delle sostanze, in opportune condizioni sperimentali può, più o meno facilmente, assumere lo stato colloidale.
Classificazione
I colloidi a seconda dello stato di aggregazione (solido S, liquido L, gassoso G) della fase dispersa e del mezzo disperdente possono essere così classificati (la prima lettera si riferisce allo stato di aggregazione della fase disperdente, la seconda della fase dispersa): S-S: sol solidi; S-L: emulsioni solide; S-G: schiume solide; L-S: sol; L-L: emulsioni; L-G: schiume; G-S: aerosol solidi; G-L: aerosol liquidi. Le particelle dei colloidi vengono evidenziate mediante osservazione all’ultramicroscopio mentre la forma può venire individuata all’esame col microscopio elettronico. Essa può essere di tipo sferoidale (ovoalbumina, emoglobina ecc.) e di tipo lineare (cellulosa, gomma, poliammidi, polistireni ecc.). Ogni particella è costituita da un numero variabile di molecole, ciascuna delle quali costituisce un’unità strutturale e funzionale.
I sistemi c. con mezzo disperdente liquido vengono generalmente distinti in liofili e liofobi a seconda del comportamento fisicochimico della fase dispersa nei riguardi del mezzo disperdente (se è acqua, si parla di colloidi idrofili e idrofobi). I colloidi liofili, detti anche emulsoidi, si formano quando la fase dispersa (sostanze solide o liquide, quali saponi, gelatine, amido) presenta una certa affinità con il mezzo disperdente da cui viene solvatata. I colloidi liofobi si possono ottenere soltanto in liquidi nei quali la sostanza da disperdere è insolubile o pochissimo solubile; viene quindi a mancare la solvatazione, responsabile dell’unione attraverso legami della fase dispersa con il liquido, e la dispersione deve essere effettuata in presenza di una sostanza peptizzante (➔ peptizzazione). I colloidi liofili si suddividono a loro volta in molecolari e micellari; le particelle nei primi sono macromolecole, nei secondi, vengono chiamate micelle e sono composte da più molecole, in genere di piccola massa, tenute insieme da legami deboli. Fra le sostanze più comuni che si trovano allo stato c. vi sono le proteine, i polisaccaridi, le gomme (colloidi liofili molecolari), i saponi (colloidi liofili micellari), i sol dei metalli (colloidi liofobi).
Cambiamenti di stato e stabilità
I colloidi possono presentare due importanti cambiamenti di stato, la flocculazione e la gelificazione. La prima consiste nella riunione di più particelle destabilizzate in modo di formare aggregati che precipitano sotto forma di polvere o di precipitato fioccoso o spugnoso; può essere reversibile o irreversibile, a seconda della possibilità o meno del ritorno allo stato di dispersione del precipitato a contatto col mezzo disperdente; generalmente i colloidi liofili sono reversibili, i liofobi irreversibili; la reversibilità non è una proprietà assoluta e altri fattori possono influire su di essa. La gelificazione consiste invece nella solidificazione del sistema in una massa apparentemente elastica ed omogenea detta gel.
La stabilità delle dispersioni c. è essenzialmente dovuta all’adsorbimento di cationi o anioni provenienti dalla soluzione, che conferiscono una carica elettrica alla superficie delle particelle disperse. Un esempio viene offerto dalle particelle di silice disperse in acqua, che danno origine superficialmente ad acido silicico, il quale subisce a sua volta un’ulteriore dissociazione ionica:
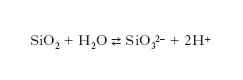
Sulla superficie delle particelle vengono così segregate cariche negative che esercitano un’azione attrattiva sugli idrogenioni presenti nella soluzione, con formazione di un doppio strato elettrico. L’adsorbimento di ioni inorganici od organici produce una barriera di energia elettrostatica che impedisce alle particelle di coagulare. Il modello per descriverne il comportamento viene contraddistinto con l’acronimo DLVO (da B. Derjagin, L.D. Landau, E.J.W. Verwey e J.Th. Overbeek). Se le particelle sono sufficientemente grandi, in prima approssimazione la loro interazione può essere espressa in funzione della distanza h fra due superfici parallele. Le particelle si respingono per effetto delle loro cariche elettrostatiche schermate dagli ioni presenti nella soluzione, attraverso un potenziale VR(h) e si attraggono per effetto di un potenziale VA(h) dovuto alle forze di van der Waals. Queste si manifestano fra i diversi atomi presenti nelle particelle e devono essere sommate per tutti gli atomi che le costituiscono. Globalmente il potenziale di interazione fra due particelle risulta descritto da una funzione data dalla somma di entrambi i contributi, attrattivo e repulsivo:
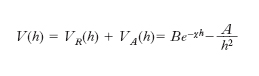
La costante A, dipende dalla natura del materiale presente nelle particelle e ha un valore intorno a 10–20-10–19 J. La presenza di un mezzo disperdente, invece del vuoto, abbassa il valore dell’energia attrattiva e se ne può tener conto modificandola opportunamente. L’andamento dell’equazione precedente è rappresentato nella figura. L’energia presenta un massimo e quindi scende verso valori negativi che portano alla formazione di un agglomerato fra due particelle. Perché ciò avvenga è necessario superare una barriera di energia potenziale la cui entità dipende dal valore dei diversi parametri che compaiono nella relazione precedente e che sinteticamente sono stati compendiati nel parametro B per quanto concerne la parte repulsiva. Di particolare interesse è il ruolo del parametro χ che dipende dal prodotto fra la concentrazione degli ioni in soluzione e il quadrato della loro carica. All’aumentare di χ diminuisce l’altezza della barriera di potenziale e pertanto risulta agevolato il passaggio verso la configurazione più stabile che porta alla coagulazione delle particelle.
Proprietà
Le proprietà chimico-fisiche dei sistemi c. dipendono dalla grandezza, forma, struttura e stato di dispersione delle particelle. Mentre non sono misurabili né l’abbassamento crioscopico, né l’innalzamento ebullioscopico, è invece agevole la determinazione della pressione osmotica, poiché sono disponibili membrane permeabili al solvente, ma non al soluto. La diffusività e la velocità di una particella c. sono grandezze facilmente determinabili dai cui valori è stato possibile determinare il peso delle particelle c. mediante l’impiego dell’ultracentrifuga. Mentre al comune microscopio le soluzioni c. appaiono otticamente vuote, la presenza di particelle c. viene evidenziata dall’effetto Tyndall, secondo il quale, se l’indice di rifrazione delle particelle è diverso da quello del mezzo disperdente, osservando il mezzo ortogonalmente alla direzione di un forte fascio luminoso incidente, il percorso del fascio stesso appare nella forma di un cono luminoso. Applicando un campo elettrico, le particelle c. si orientano in esso e migrano (elettroforesi), confermando la presenza di cariche superficiali su di esse. Quantitativamente la caratterizzazione elettroforetica di una particella avviene attraverso il valore della mobilità elettroforetica.
Azione
I colloidi hanno grande importanza sia dal punto di vista scientifico (studio e comprensione di molti fenomeni biologici), sia da quello applicativo, nell’industria (tessili, materie plastiche, gomme, tensioattivi) e in agricoltura (humus, generi alimentari).
Colloide protettore Sostanza che migliora la stabilità di un sistema eterogeneo o di una dispersione c.; per es. una piccola quantità di caseina, aggiunta a una soluzione colloidale d’oro, impedisce che questa coaguli in presenza di un elettrolita quale il cloruro di sodio. L’azione dei colloidi protettori è dovuta a fenomeni di adsorbimento in seguito ai quali le particelle del sistema colloidale instabile si rivestono del c. protettore che le rende meno sensibili all’azione degli elettroliti.
Presenza in natura
I colloidi sono presenti in natura nelle strutture degli organismi animali e vegetali e nel terreno, o costituiscono liquidi organici naturali, come sangue e siero delle piante.
In particolare si dà il nome di colloide tiroidea alla sostanza gelatinosa, di aspetto omogeneo, presente nell’interno delle vescicole o follicoli della ghiandola tiroide. Contiene l’ormone tiroideo, denominato tiroxina, legato a una proteina del tipo delle globuline a formare la tireoglobulina, che rappresenta la forma chimica sotto la quale l’ormone è immagazzinato nella cavità delle vescicole tiroidee. Contiene inoltre alcune mucoproteine e parecchi enzimi.
Per la colloidoclasia, turbamento dell’equilibrio colloidale del plasma sanguigno ➔ emoclasia.
Abstract di approfondimento da Colloidi di Silvia Ardizzone (Enciclopedia della Scienza e della Tecnica)
Nel 1907, Wolfang Ostwald definì i sistemi colloidali «il mondo delle dimensioni trascurate», ponendo l’accento sul fatto che le dimensioni dei componenti di tali sistemi non hanno né l’estensione tipica delle molecole, né quella dei materiali cristallini. La definizione generale di colloide poggia ancora oggi sulla dimensione lineare dei suoi componenti – compresa tra 1 nm e 1 mm – considerata la caratteristica fondamentale delle particelle che lo compongono. Per questa ragione, lo studio dei colloidi è fortemente interdisciplinare e abbraccia la chimica, la fisica, la biologia, la scienza dei materiali, e molte altre scienze. La chimica dei colloidi è dunque la scienza sia delle grandi molecole, sia dei sistemi multifase in stato di elevata suddivisione. Nello studio di tali sistemi si incontrano la scienza dei colloidi e quella delle interfasi: infatti, tanto più una certa fase è suddivisa, tanto maggiore è l’estensione di area superficiale a parità di peso del campione, e quindi tanto più rilevanti sono i fenomeni che si verificano al contatto tra la fase suddivisa e le altre fasi (interfase).
Non esiste ramo dell’industria chimica che non coinvolga proprietà o metodologie dei colloidi e delle interfasi. Inoltre, sistemi nell’intervallo dimensionale dei colloidi si incontrano regolarmente nelle scienze ambientali (aerosol, nebbie, schiume, ecc.), nella scienza dei materiali (leghe, cementi, fibre, ecc.) e nei prodotti per uso domestico e cosmetico (latte, maionese, ecc.). Molte sospensioni (vale a dire dispersioni di solidi nei liquidi), come le vernici, gli inchiostri, le lacche, gli anticrittogamici, ecc., sono di uso comune; altre, come per esempio le sospensioni di minerali in acqua, che si prestano al trasporto in condotta, lo diverranno presto. Altri tipi di sistemi, come le schiume strutturali e, in senso lato, i materiali compositi, sostituiranno tra breve i materiali ferrosi e le leghe di alluminio; altri ancora – per esempio, i liposomi, le micelle o le emulsioni – sono già entrati nella preparazione di alcuni farmaci, potenziandone enormemente le proprietà. A testimonianza della grande ;importanza dei colloidi potremmo citare numerosi altri esempi, come quelli biologici (sangue, cellule, lecitine o fosfolipidi di membrana).
Storicamente, la scienza dei colloidi nasce nel 1845 con l’opera di Francesco Selmi, il quale descrisse alcune «strane proprietà» di soluzioni in acqua di zolfo, cloruro di argento e blu di Prussia. Nel 1861, Thomas Graham coniò il termine colloide – che deriva dalla parola greca kálla e significa proprio materiale colloso – per indicare un insieme amorfo e mal definito. Graham riteneva che esistessero due stati diversi della materia con proprietà chimico-fisiche completamente differenti, i cristalloidi e appunto i colloidi, assimilabili i primi ai minerali e i secondi a una massa organizzata. In realtà, a seconda della specifica applicazione del colloide, le loro condizioni di preparazione possono essere modulate in modo da privilegiare alternativamente l’estensione del contatto interfasale o la crescita dei cristalliti. Tuttavia, le proprietà di maggiore interesse dei sistemi colloidali si individuano nel comportamento del sistema nel suo complesso (reologia, proprietà ottiche, stabilità) e, nel caso dei sistemi multifase, nello stato superficiale e nella reattività interfasale.
Classificazione dei sistemi colloidali
Possono essere considerati colloidi sia le grandi molecole, sia i materiali (i solidi, i liquidi e anche i gas) in stato di elevata suddivisione, a patto che le componenti del sistema (molecole o particelle) abbiano dimensioni comprese tra 1 nm e 1 mm. La differenza tra questi due tipi di colloidi è collegata alla relazione esistente tra la particella colloidale e il mezzo in cui essa si trova. Per esempio, i colloidi macromolecolari danno luogo a vere soluzioni – nel senso termodinamico del termine, ossia attraverso un processo spontaneo – con il mezzo che li circonda. Una fase suddivisa, d’altra parte, forma con il mezzo un sistema almeno bifasico, con la presenza di un confine di fase esteso. I sistemi colloidali monofasici sono definiti liofili, quelli bifasici liofobi. I colloidi liofili possono formare vere soluzioni, che si creano spontaneamente quando soluto e solvente sono messi in contatto. In assenza di cambiamenti chimici o di temperatura, un sistema colloidale liofilo è stabile indefinitamente. Viceversa, quando due fasi sono portate in contatto, esse non danno origine spontaneamente a una dispersione finemente suddivisa, anzi, se si lascia riposare il sistema, si instaura il processo opposto: per esempio, olio e acqua possono essere mescolati vigorosamente per produrre un fluido eterogeneo e non trasparente, ma, a riposo, il sistema evolverà verso due strati omogenei e trasparenti.