
imprinting genètico
imprinting genètico (o genomico) Forma di eredità epigenetica nella quale, durante la formazione dei gameti, viene modificato il livello di espressione di un gene o di un cromosoma.
Caratteri generali
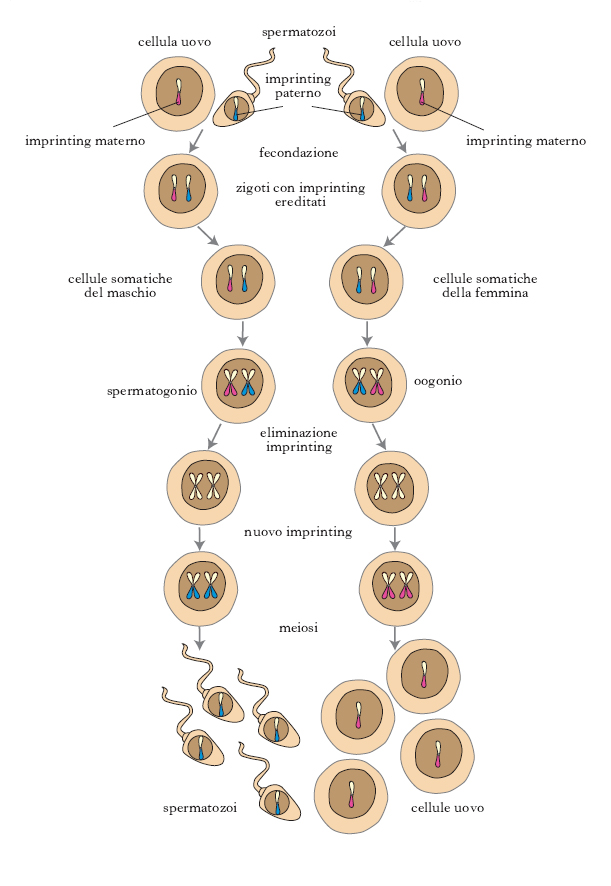
Le cellule somatiche dello zigote serbano la memoria delle modificazioni avvenute in uno dei gameti parentali e sono pertanto marcate (imprinted). L’i. condiziona l’espressione del gene paterno o materno in quell’individuo, ma non nei discendenti. Sulla base dei principi mendeliani, dato che ciascun genitore fornisce alla progenie metà del numero dei cromosomi, si è creduto per lungo tempo che entrambi i genitori dessero un uguale contributo genetico, cioè che l’espressione di un certo carattere non dipendesse dal sesso del genitore dal quale esso derivava. Gli studi degli anni 1990 hanno dimostrato che alcuni geni, o tratti di cromosomi, sono invece imprinted, per cui alleli ereditati dal padre sono espressi in maniera diversa da quelli corrispondenti ereditati dalla madre (fig. 1).
Prove convincenti dell’i. genetico derivano dagli studi delle prime fasi di sviluppo degli embrioni murini. Poiché nelle cellule uovo fecondate di topo è facile distinguere il pronucleo maschile da quello femminile, sperimentalmente si può rimuovere uno dei due pronuclei, per es. quello paterno, e se ne può inserire un altro di origine materna. Si formano così embrioni con genomi solo materni, detti ginogenetici, che si accrescono più o meno regolarmente per un certo tempo senza sviluppare però le strutture extraembrionali. Se sono presenti invece due genomi paterni, l’embrione (androgenetico) non si forma, anche se vi sono le membrane extraembrionali. Dal momento che nessuna di queste situazioni è compatibile con uno sviluppo embrionale e fetale normale, risulta evidente che i due genomi danno contributi necessari e funzionalmente distinti all’embriogenesi. Sono inoltre note due condizioni patologiche umane che confermano che l’assenza di geni di uno dei due genitori non consente uno sviluppo normale: la mola idatiforme, tumore placentale umano dovuto alla presenza di due corredi cromosomici paterni, senza contributo materno; i teratomi ovarici, tumori benigni che contengono tessuti ben differenziati e derivano da cellule che presentano solo cromosomi materni.
Numerose evidenze dimostrano che l’espressione differenziale dei geni nel fenomeno dell’i. è collegata alla metilazione del DNA, causa di inattivazione della trascrizione durante la gametogenesi maschile e femminile. In modelli sperimentali, quali topi transgenici in cui è stata inattivata la DNA-metiltransferasi, si è osservata la morte precoce degli embrioni in conseguenza della riduzione del 95% della metilazione. Questo esperimento, pur essendo molto significativo, non dimostra tuttavia se la metilazione sia la causa dell’i. o se mantenga semplicemente lo stato imprinted del gene una volta che il fenomeno si sia verificato. Le ricerche compiute sul genoma murino hanno dimostrato che esistono almeno 10 regioni, distribuite su 6 diversi cromosomi, nelle quali gli alleli paterni e materni sono espressi differentemente.
Analisi molecolare di malattie genetiche
Le ricerche nell’uomo dimostrano che l’espressione fenotipica di alcune malattie è diversa a seconda che esse siano ereditate dal padre o dalla madre. Le maggiori differenze cliniche si notano nelle due sindromi ereditarie note come sindrome di Prader-Willi e sindrome di Angelman. Nella sindrome di Prader-Willi, caratterizzata da obesità, bassa statura, ipogonadismo, ritardo mentale e ipotonia, si osserva frequentemente una delezione di un tratto del braccio lungo del cromosoma 15. Il cromosoma 15 deleto è ereditato dal padre; pertanto l’individuo affetto dalla malattia ha, per quel tratto di cromosoma, informazioni genetiche esclusivamente materne. È stato dimostrato che, se la delezione dello stesso tratto di cromosoma 15 è ereditata dalla madre, i pazienti presentano invece la sindrome di Angelman, completamente diversa dalla precedente, caratterizzata da movimenti atassici, simmetrici e ripetitivi, attacchi convulsivi e accessi improvvisi di risa. Questo suggerisce l’esistenza di due geni, l’uno con espressione paterna e l’altro materna, che codificano distinte funzioni. Dalla fine degli anni 1980 sono stati descritti vari individui, affetti dalla sindrome di Prader-Willi, che presentano cromosomi 15 normali senza delezioni. Mediante l’uso di sonde molecolari si è potuto capire che essi avevano entrambi i cromosomi di origine paterna. Questa situazione inusuale, chiamata disomia uniparentale, dimostra che, per uno sviluppo normale, i geni di questa regione cromosomica devono essere ereditati in doppia copia, ma ciascun membro della coppia deve derivare da un genitore diverso. Una spiegazione verosimile è che in quest’ultimo caso si sia inizialmente verificata una trisomia del cromosoma 15, a seguito di anomalie della meiosi paterna o materna, poi compensata dalla perdita di uno dei cromosomi soprannumerari. Analizzando il locus della delezione sul cromosoma 15 si è scoperto che la delezione interessa una serie di geni: alcuni di essi trascrivono RNA non codificanti, mentre il gene SNRPN codifica la proteina SmN, costituente fondamentale di una delle particelle snRNP (small nuclear ribonucleoprotein) coinvolte nello splicing di RNAm specifici nelle cellule del cervello. Questa scoperta suggerisce una forte correlazione fra la mutazione di SNRPN e i seri disturbi comportamentali degli individui affetti da questa sindrome. È stato identificato, 450 chilobasi a valle di SNRPN, il gene responsabile della sindrome di Angelman, UBE3A, che codifica una proteina che lega l’ubiquitina. Nel 1997 è stata dimostrata, da parte di U. Albrecht e collaboratori, l’espressione materna del gene nelle cellule del cervello. Dato che alcuni geni, distanti diverse migliaia di kilobasi dalle zone di DNA coinvolte nelle due sindromi, subiscono ancora l’influsso dell’i., si pensa che vi possano essere due lunghe regioni di controllo dell’i. che interagiscono fra loro; a queste regioni è stato dato il nome di PWS-ICR (Prader-Willi syndrome imprinting control rregion) e AS-ICR (Angelman syndrome imprinting control region).
Tratti di genoma suscettibili di i. possiedono un centro di inattivazione che durante la maturazione delle cellule germinali agisce attraverso una metilazione di dinucleotidi CpG; sotto l’influsso del centro di inattivazione viene modificato un intero tratto di cromatina, con il risultato che il dispositivo di trascrizione non ha più accesso ai geni interessati, come avviene nella regione di controllo di locus (LCR) della famiglia genica delle globine (➔ globina); non si sa tuttavia come il centro di inattivazione possa influenzare la struttura di un lungo tratto di cromatina.
L’i. è presente nei Mammiferi ma non negli altri Vertebrati e ci si domanda pertanto quale possa essere il suo vantaggio evolutivo. Probabilmente, sia il controllo della crescita del feto sia le relazioni materno-fetali sono stati le forze selettive che hanno agito sull’instaurarsi dell’i. nei Mammiferi, lo sviluppo dei quali è essenzialmente caratterizzato dalla presenza della placenta, che permette le interazioni fra l’embrione e la madre. Particolarmente interessanti sono le analogie che l’i. presenta con l’inattivazione del cromosoma X (➔ epigenesi) e che si possono così riassumere: il meccanismo inattiva uno dei due cromosomi di una cellula diploide; l’inattivazione avviene specificamente nel cromosoma X paterno a livello del tessuto placentare ed è invece casuale nei tessuti embrionali; il prodotto di XIST, un gene coinvolto nell’inattivazione del cromosoma X, così come il prodotto di alcuni geni coinvolti nell’i. del topo, è un RNA non codificante che esplica la sua azione sui geni localizzati sullo stesso cromosoma; la metilazione, essenziale per l’i., è anche il meccanismo che regola l’espressione di XIST.