
Polimeri
Polimeri
Il termine polimero (dal greco poly-, molte, e méros, parte) vuol dire costituito di molte parti, ed è stato usato in passato nella scienza con significati affini, ma non sempre coincidenti. In questo saggio ci atterremo alle definizioni elaborate dalla Commissione di nomenclatura macromolecolare della IUPAC (International Union of Pure and Applied Chemistry).
Un polimero è una sostanza composta da molecole caratterizzate dalla ripetizione multipla di unità strutturali chimiche, costituite da una o più specie di atomi o gruppi di atomi legati tra loro (unità costituzionali, nella terminologia IUPAC), in modo da formare catene di atomi. Un monomero (dal greco mónos, singolo, e méros, parte) è un composto che consiste di molecole tutte uguali, ciascuna delle quali può fornire un'unità strutturale di ripetizione nel polimero detta unità monomerica. Un polimero si dice regolare se le molecole da cui è costituito possono essere descritte da una sola specie di unità strutturali, in una singola disposizione sequenziale. Numerosi polimeri naturali e sintetici sono polimeri regolari, come la cellulosa (naturale), la cui unità strutturale deriva dal glucosio, e il polietilene (artificiale), la cui unità strutturale deriva dall'etilene, di formula CH2=CH2 che può essere scritto come:
−(CH2−CH2)−n, con n≫100.
Se nelle catene polimeriche vi sono atomi di carbonio tetraedrici legati a due sostituenti diversi, per esempio R e H,
H
∣
catena−C−catena
∣
R
(come avviene nella cellulosa) o trigonali e collegati da un doppio legame, come, per esempio, in
catena−CH=CH−catena
i collegamenti possono avvenire, in tre dimensioni, in due modi stericamente (cioè spazialmente) diversi. Un polimero si dice stereoregolare se tali collegamenti avvengono sempre secondo una ben precisa disposizione sequenziale.
L'ammontare di unità strutturali (unità monomeriche) legate tra loro nelle molecole di un polimero deve essere sufficientemente grande da dare al polimero un insieme di proprietà che non variano marcatamente per aggiunta o sottrazione di più unità strutturali alle sue molecole. Il numero di unità monomeriche contenute nelle molecole di un polimero è detto grado di polimerizzazione; di tale grandezza si può definire un valore medio attraverso appropriate misure fisiche. Normalmente, esso è dell'ordine di ben oltre il centinaio sia nei polimeri sintetici sia in quelli naturali. Per questo, Hermann Staudinger propose nel 1920 di chiamare macromolecole (mácros in greco vuol dire grande, esteso) le molecole contenute in un polimero. La massa molecolare media delle macromolecole contenute in un polimero (detta ancora comunemente, ma impropriamente, peso molecolare del polimero) è il prodotto del grado di polimerizzazione per la massa molecolare (peso molecolare) dell'unità monomerica.
La struttura chimica dei polimeri
Già nel 1920 Staudinger contestò le teorie correnti sulla natura delle sostanze polimeriche, considerate composti di associazione tenuti insieme da valenze secondarie, proponendo per i polimeri sintetici dello stirene, della formaldeide e per la gomma naturale le formule a catena aperta attualmente in uso. Staudinger attribuì le proprietà colloidali degli alti polimeri esclusivamente all'elevato peso molecolare delle loro molecole, che, appunto, propose di chiamare macromolecole.
Tali concetti non furono subito accettati e la polemica che ne seguì continuò per tutti gli anni Venti del Novecento. Le teorie degli oppositori, però, finirono col cadere di fronte a nuove dimostrazioni sperimentali, quali quelle derivanti dall'indagine con i raggi X di vari polimeri cristallini e dai lavori di sintesi di Wallace Carothers. Questi preparò numerosi polimeri (in particolare, come vedremo più avanti, il nylon) attraverso semplici reazioni organiche, che non lasciavano dubbi circa la struttura lineare delle macromolecole ottenute.
Il chiarimento del concetto di macromolecola pose su basi scientifiche lo sviluppo della chimica che studiava queste sostanze. Il periodo che va dalla metà degli anni Trenta agli inizi degli anni Cinquanta vide uno sviluppo rapidissimo di questa scienza, in tutti i suoi aspetti. Furono poste le basi, teoriche e pratiche, per la determinazione dei pesi molecolari medi e fu chiarita la costituzione dei più importanti polimeri. Contemporaneamente, veniva chiarito, negli aspetti essenziali, il meccanismo chimico della polimerizzazione, che può avvenire a stadi (cioè per successive condensazioni, come nel caso del nylon) o a catena (cioè per successive addizioni). Ciò portò a distinguere, nella polimerizzazione a catena, tra quella radicalica, quella cationica e quella anionica. Venne anche affrontato lo studio della copolimerizzazione, cioè della polimerizzazione, nella stessa catena, di due o più monomeri diversi, e ne furono chiarite le leggi fondamentali.
I problemi connessi con la conformazione delle macromolecole cominciarono a essere affrontati fin dagli anni Trenta da grandi scienziati come Herman Francis Mark e Werner Kuhn. Da allora furono poste le basi teoriche per affrontare i problemi relativi al comportamento dei polimeri in soluzione e all'elasticità della gomma. Vennero anche affrontati i problemi riguardanti i vari possibili stati fisici delle sostanze polimeriche e furono chiarite, negli aspetti essenziali, le relazioni tra proprietà e struttura.
Con i raggi X fu visto che numerosi polimeri di origine naturale, quali la cellulosa, le proteine fibrose, la guttaperca, sono cristallini; la gomma naturale cristallizza facilmente per raffreddamento o sotto stiro. Numerosi poli-meri sintetici sono pure cristallini. In generale, i diagrammi di diffrazione a raggi X dei polimeri cristallini sono più diffusi e meno ricchi di riflessioni di quelli dei composti a basso peso molecolare; le differenze indicano dimensioni più piccole delle zone cristalline e imperfezioni all'interno di esse.
L'aspetto che caratterizza lo stato cristallino è l'ordine tridimensionale a lunga distanza. Nei cristalli di sostanze di massa molecolare piccola, l'ordine tridimensionale risulta dalla sistemazione precisa di molecole identiche o al più enantiomorfe, in posizioni equivalenti di ogni cella elementare e dalla ripetizione identica in tre dimensioni di tali celle elementari, per dar luogo al reticolo cristallino.
Nel caso dei polimeri, i siti reticolari dei cristalli sono occupati da piccole unità chimiche che si ripetono lungo le catene delle macromolecole e che sono connesse da legami covalenti. Ciò apparve evidente fin dai primi studi sull'argomento riguardanti i polimeri naturali.
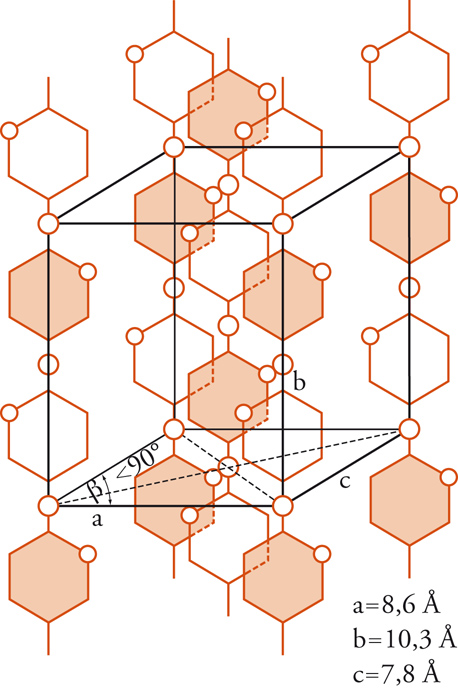
Un modello schematico, proposto nel 1928 per la cellulosa da Kurt Meyer e da Mark, è mostrato nella fig. 2. Come si vede, nella cella elementare della cellulosa le unità strutturali ripetitive giocano un ruolo simile a quello delle molecole singole nel caso di sostanze di massa molecolare piccola: le macromolecole passano da una cella all'altra attraverso tutto il reticolo cristallino lungo l'asse diretto in senso verticale nella figura.
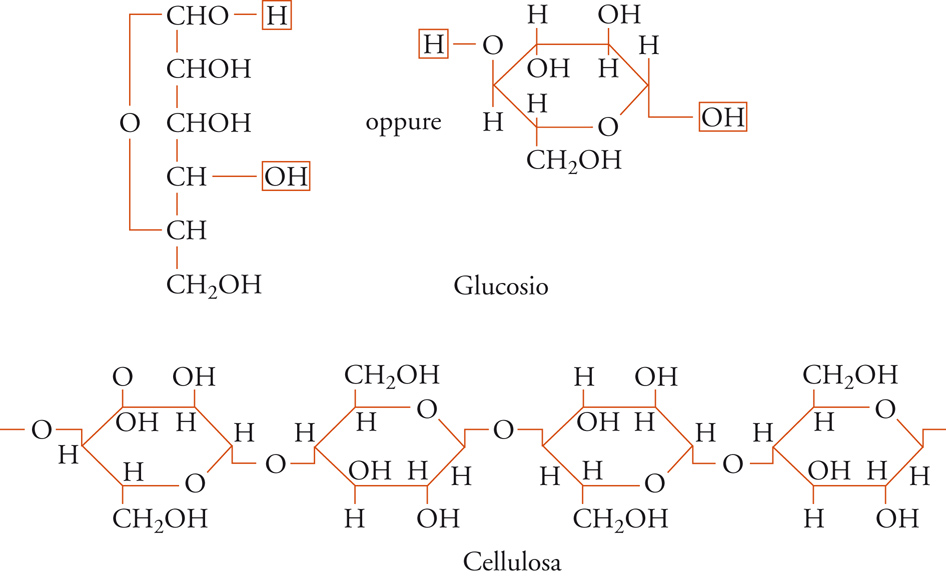
L'uguaglianza delle unità strutturali lungo la catena di un polimero cristallino può essere garantita se è regolare la struttura chimica del polimero stesso: per esempio, è stato visto che lungo la catena della cellulosa si succedono unità tutte di β-glucosio e tutte in concatenamento 1-4 (fig. 3); lungo la catena dell'amido (in particolare della sua frazione solubile in acqua) si succedono unità tutte di α-glucosio e tutte in concatenamento 1-4.
Come abbiamo visto in precedenza, attualmente si definisce polimero regolare un polimero lineare le cui molecole possono essere descritte sostanzialmente da una sola specie di unità costituzionale, in un singolo arrangiamento sequenziale. Definiamo un polimero stereoregolare se è regolare anche la successione di configurazioni (intendendo per configurazione la disposizione spaziale dei legami, prescindendo tuttavia dalla molteplicità delledisposizioni spaziali che possono sorgere per rotazioneattorno a legami semplici). Cellulosa, amilosio, guttaperca e gomma naturale sono esempi di polimeri stereoregolari che la natura è capace di sintetizzare.
3. Ordine e/o disordine nella successione di configurazioni

Nei polimeri ottenuti per sintesi, l'ordine nella struttura chimica delle macromolecole può essere impedito da numerosi tipi di irregolarità. Le costituzioni chimiche delle unità che si succedono lungo la catena possono essere differenti, come nel caso, abbastanza frequente, che dalla molecola di un certo monomero (nel caso mostrato nella fig. 4 a sinistra, il butadiene), si possano ottenere due o più unità costituzionali diverse.
Anche nel caso in cui le costituzioni chimiche delle unità che si susseguono lungo la catena di un polimero siano tutte uguali (fig. 4 a destra), la loro struttura può essere polare nel senso che i due terminali delle unità sono non equivalenti. Le unità monomeriche derivabili da un monomero vinilico (per es., il propilene) sono illustrative al riguardo. Il disordine che si può immaginare è nel collegamento delle unità denominato testa-testa oppure testa-coda.
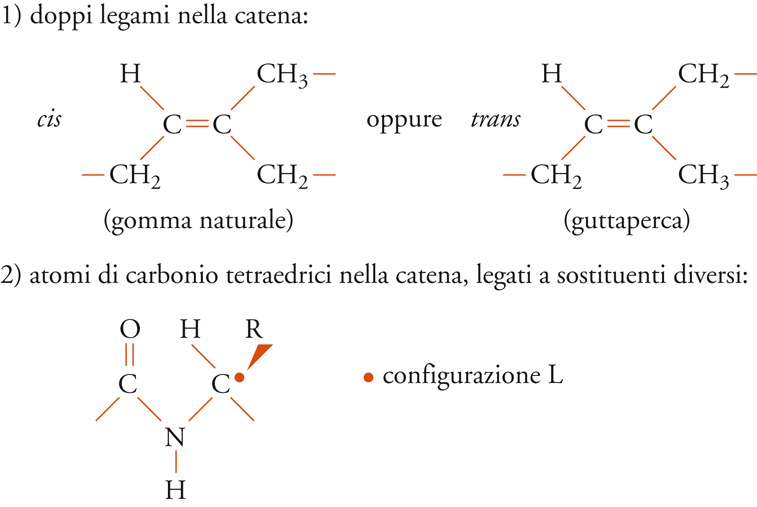
Infine, le unità possono avere la stessa costituzione e lo stesso orientamento polare, ma differire nella configurazione. Ciò può accadere ogni qualvolta lungo la catena si abbia un doppio legame, oppure un atomo di carbonio tetraedrico legato a due sostituenti diversi (fig. 5). Il primo caso è ben illustrato dalle unità monomeriche dei due poliisopreni naturali (guttaperca e gomma naturale), il secondo riguarda sia i polimeri vinilici che la maggior parte dei biopolimeri (ordine strutturale). Nel caso dei biopolimeri, per esempio la fibroina della seta o la cellulosa (fig. 3), le configurazioni degli atomi di carbonio tetraedrici (asimmetrici) sono notevolmente regolari per ogni macromolecola (anche se le singole macromolecole differiscono l'una dall'altra per massa molecolare, come nei polimeri sintetici).
L'arrangiamento sequenziale delle unità strutturali è precisamente definito nelle proteine, che sono composte da una ventina di residui di amminoacidi, e nei polinucleotidi, che risultano dal concatenamento di quattro unità fondamentali (nucleotidi). Come già si è detto, le corrispondenti (macro)molecole, per ciascuna specie, sono uniformi, hanno cioè tutte la stessa massa molecolare.
Le chiralità ‒ destra o sinistra ‒ degli atomi di carbonio legati a quattro sostituenti diversi, che sono presenti in grandi quantità in queste molecole, sono unicamente specificate, così come sono unicamente specificati i centri asimmetrici presenti nelle unità strutturali della cellulosa e di altri polisaccaridi.
L'ottenimento per sintesi di copolimeri da una miscela di molti monomeri, con un concatenamento corrispondente a una sequenza prescritta come nelle proteine native, sarebbe un'impresa ben al di là delle possibilità attuali dei metodi sintetici di cui disponiamo.
E, purtuttavia, come vedremo, principalmente attraverso le ricerche di Giulio Natta e della sua scuola, la regolazione della struttura chimica e stereochimica è possibile per numerosi polimeri sintetici. La cristallinità che può essere ottenuta per tali polimeri è la chiave per il conseguimento di un insieme di proprietà d'uso altrimenti non ottenibile.

Facciamo riferimento ai polimeri vinilici, vale a dire a quei polimeri ottenibili da monomeri vinilici, CH2=CHR, quali il propilene, lo stirene, il cloruro di vinile. A tale proposito, per quanto concerne questi ultimi polimeri, si può notare che se abbiamo un composto con R=H, e si apre il doppio legame per far concatenare le molecole del monomero ‒ che in tal caso sarebbe l'etilene, CH2=CH2 ‒ c'è un solo modo di concatenare le unità monomeriche (fig. 6).
Se si prende in considerazione, invece, la polimerizzazione del propilene in cui R=CH3, o del cloruro di vinile, R=Cl, ogni volta che si aggiunge alla catena un'unità monomerica, ci sono quattro modi diversi di farlo. N unità viniliche, se entrassero in una catena casuale con concatenamento primario oppure secondario, e ancora a caso con le due possibili diverse chiralità, potrebbero succedersi in quattro modi diversi. Le connessioni in una macromolecola di N unità monomeriche potrebbero effettuarsi in 4N=10N log4=100,6N modi diversi.
Per esempio, una singola macromolecola di 1000 unità monomeriche potrebbe realizzarsi per sintesi, in assen-za di un meccanismo ordinatore, in 41000=10600 modidiversi. Per avere un'idea dell'ordine di grandezza di tale numero, si noti che l'ordine di grandezza del numero di atomi presenti nell'Universo è stimato attualmente in 1082. Come direbbe Dante delle infinite scintille angeliche: "Ed eran tante che il numero loro/più che il doppiar degli scacchi s'immilla" (Paradiso, XXVIII, 92-93).
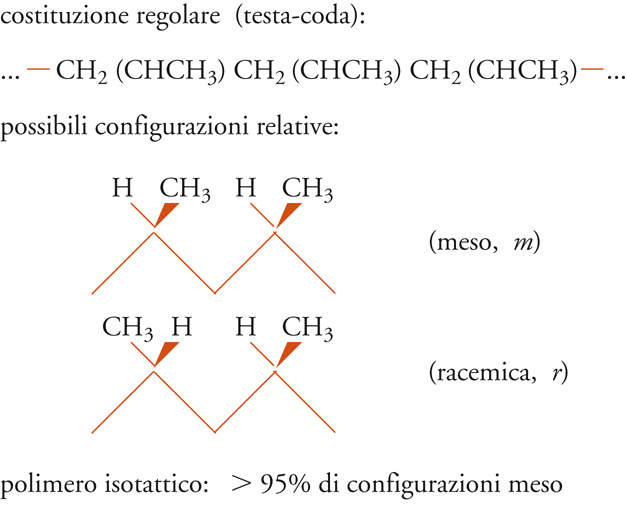
Ritornando ai polimeri vinilici, la loro costituzione è definita regolare, se l'arrangiamento sequenziale delle unità monomeriche è prevalentemente del tipo testa-coda, come è evidenziato in alto, nello schema della fig. 7, nel caso dei polimeri del propilene.
Questo tipo di ordine strutturale non è però sufficiente, in generale, a conferire ordine tridimensionale e quindi cristallinità a tali polimeri, fintantoché manca l'ordine nella successione delle configurazioni degli atomi di carbonio terziari. La catena è regolare ma non è ‒ come si dice ‒ stereoregolare.
Se, infatti, consideriamo una porzione della catena polimerica nella sua conformazione estesa, zig-zag planare, vediamo chiaramente che si possono avere due diverse configurazioni relative degli atomi di carbonio terziari di una coppia (o diade) di unità monomeriche adiacenti.
Nel caso delle diadi meso, i gruppi CH3 si trovano dalla stessa parte rispetto al piano della catena distesa nella conformazione zig-zag planare; nel caso delle diadi racemiche, i gruppi CH3 sono da parti opposte rispetto al piano della catena distesa nella conformazione zig-zag planare.
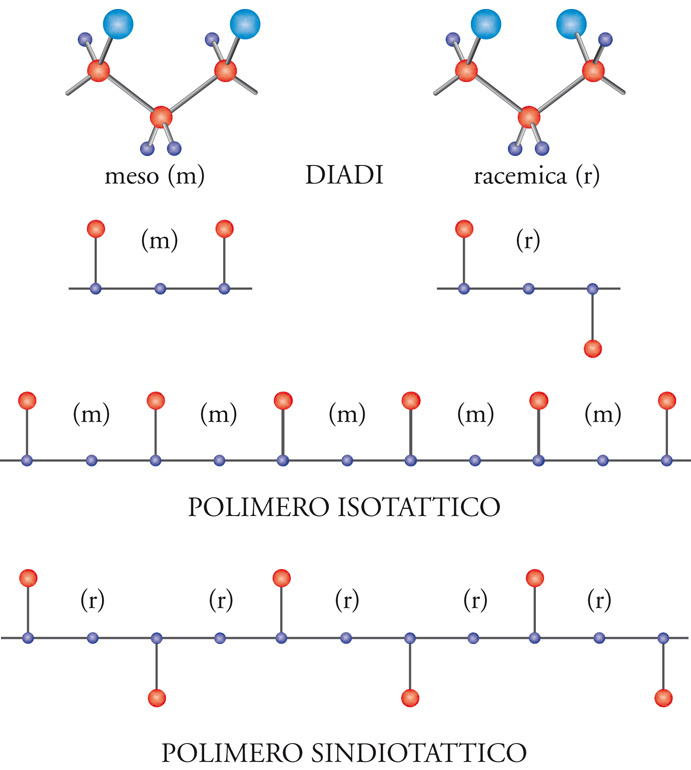
In definitiva, è possibile ottenere l'ordine strutturale chimico e stereochimico, che è un prerequisito alla cristallizzabilità di un polimero vinilico, con un concatenamento testa-coda delle unità monomeriche, per quanto riguarda l'ordine chimico; con una successione di diadi tutte meso oppure con una successione di diadi tutte racemiche per quanto riguarda l'ordine stereochimico (fig. 8).
Nel primo caso si ha un polimero isotattico (in cui, cioè, le unità successive hanno tutte la stessa configurazione sterica); nel secondo caso, un polimero sindiotattico (in cui, le unità successive hanno configurazione sterica opposta). Come vedremo più avanti, il concetto di configurazione è entrato nella scienza dei polimeri con la scoperta del polipropilene isotattico e i conseguenti studi della scuola di Natta.
4. Il sorgere di un'industria dei polimeri artificiali e sintetici
Prima della fine della Prima guerra mondiale e prima che le idee fortemente propugnate da Staudinger si affermassero (nel periodo 1918-1939), pochi chimici accettavano l'idea che potessero esistere sostanze costituite da molecole molto grandi, macromolecole o polimeri. Eppure un'industria dei polimeri c'era già.
Nel periodo precedente le proposte di Staudinger, infatti, alcuni importanti sviluppi industriali erano basati su due principî attraverso i quali fu possibile ottenere materiali polimerici con proprietà utili per l'uomo. Il primo riguarda la modificazione chimica di macromolecole sintetizzate da organismi viventi (gomma naturale e cellulosa); il secondo l'ottenimento di un materiale modellabile in oggetti (materiale plastico) da soli prodotti chimici ottenibili per sintesi a basso costo (nella fattispecie, il fenolo e la formaldeide).
La gomma naturale (caucciù), che gli Indiani del bacino del Rio delle Amazzoni ricavavano dal lattice che sgorga da incisioni praticate sul tronco della pianta di Hevea brasiliensis, veniva usata dagli Indiani stessi per ottenerne stivali. Ne modellavano la forma direttamente sul piede, lasciando che essa si mantenesse, dopo poco tempo, sotto la blanda azione dell'aria. L'ossigeno dell'aria trasforma le macromolecole di poliisoprene 1-4 cis, di cui è costituita la gomma naturale, in una struttura reticolata, che può resistere a tensione.
Il trattamento chimico della gomma (processo di vulcanizzazione) fu inventato da Charles Goodyear nel 1844. Un esempio di vulcanizzazione da lui proposto consiste nel mescolare 100 parti di gomma con 20 parti di zolfo e 28 parti di carbonato basico di piombo, e nello scaldare la miscela a 132 °C. Precedentemente, la gomma era stata messa in commercio senza nessun previo trattamento, e usata (come accade tuttora) per cancellare tratti segnati su un foglio di carta da una matita.
Verso la fine dell'Ottocento, la gomma vulcanizzata cominciò a essere usata per la costruzione di pneumatici per le automobili e da quel momento la produzione industriale di manufatti in gomma, seguendo il destino dell'automobile, aumentò esponenzialmente. La vulcanizzazione della gomma consiste in una reticolazione assai efficiente delle macromolecole di poliisoprene 1-4 cis in essa contenute, attraverso ponti di zolfo (in luogo dei ponti di ossigeno utilizzati negli stivali degli Indiani), catalizzata da sostanze come l'ossido basico di piombo. La formulazione brevettata da Goodyear era stata trovata in modo empirico (per trial and error) attraverso un'intelligente sperimentazione tecnologica.
Tra le fibre di provenienza animale o vegetale usate dall'uomo fin dalla più remota Antichità, vi sono molti materiali polimerici con catene molecolari tutte orientate in una stessa direzione, come il cotone (fibre di cellulosa), la lana e la seta (fibre di poliamminoacidi).
Il trattamento chimico della cellulosa, che è ricavabile dal legno, fu trovato attraverso la sua trasformazione in materiale lavorabile con macchine utensili. Il trattamento con acido nitrico della cellulosa portò alla nitrocellulosa, un materiale che, benché altamente infiammabile, venne usato, plastificato con canfora, per produrre la celluloide (inventata da John W. Hyatt nel 1869); altri trattamenti, studiati tra la fine dell'Ottocento e i primi del Novecento, portarono allo sviluppo di fibre artificiali, ottenibili per estrusione, che consistevano in cellulosa (rigenerata dopo l'estrusione) o in suoi derivati chimici (per es., acetilcellulosa). Nonostante l'avvento, a partire dal nylon, delle fibre sintetiche, si ha tuttora una notevole produzione industriale di fibre artificiali ricavate dalla cellulosa.
Per quanto riguarda l'ottenimento per sintesi chimica di materiali modellabili in oggetti (materiali plastici) da soli prodotti chimici (pure ottenibili per sintesi a basso costo), è opportuno ricordare la bachelite (o bakelite), disponibile industrialmente dagli inizi del Novecento, che può essere considerata la prima materia plastica sintetica usata dall'uomo.
Il brevetto per la preparazione della bachelite fu dato a Leo Hendrik Baekeland nel 1907. La bachelite è il prodotto resinoso che si ottiene facendo reagire fenolo e formaldeide, che diventa plastico per riscaldamento; in queste condizioni può essere compresso in oggetti di varia forma. Prolungando il riscaldamento nello stampo, il materiale indurisce e mantiene permanentemente la forma che gli è stata data, perché dà luogo a una struttura chimica reticolata. La sua produzione ebbe un notevole sviluppo industriale negli anni che precedettero la Seconda guerra mondiale per produrre componenti delle radio e delle automobili.
Durante la Seconda guerra mondiale, gli statunitensi persero la possibilità di rifornirsi di gomma naturale dalle Indie Orientali. In breve tempo, essi misero a punto la produzione di una gomma sintetica basata sulla copolimerizzazione di butadiene e stirene. Essendo frutto di ricerche finanziate dal governo, la gomma prese il nome di GR-S (Government rubber-styrene) attualmente più spesso indicata con la sigla SBR (Styrene-butadiene-rubber), e tuttora prodotta in grandi quantità, dato che gli pneumatici moderni sono costruiti con materiali polimerici diversi (elastomeri), ciascuno con un'appropriata funzione ottimizzata, che ricomprende SBR, gomma naturale (ottenibile anche per sintesi) ed elastomeri sintetici come il polibutadiene 1-4 cis.
5. I primi polimeri sintetici prodotti industrialmente
Alcuni materiali polimerici, con macromolecole costituite da catene lineari, cominciarono a essere prodotti industrialmente per sintesi nel decennio 1930-1940 e la loro produzione per scopi bellici ricevette un primo impulso durante la Seconda guerra mondiale.
Macromolecole costituite da lunghe catene di atomi possono essere sintetizzate in due modi:
Polimerizzazione a catena (o per addizione), cioè per crescita continua di ogni catena, a partire da un iniziatore (per es., una molecola-radicale) fino a che non abbia luogo una reazione di terminazione, che fa cessare la crescita di quella singola catena. Nella polimerizzazione per addizione, i polimeri sono costituiti da macromolecole gigantesche che hanno la stessa composizione dei monomeri di partenza (a parte il punto di inizio o iniziazione [I] e di terminazione [T]). Iniziazione e terminazione devono essere regolate in relazione alla lavorabilità e alle proprietà d'uso del materiale che si desidera produrre. Dall'etene (o etilene), CH2=CH2, si ottengono le macromolecole che costituiscono il polietilene (noto in commercio con il nome di politene):
[I] formula.
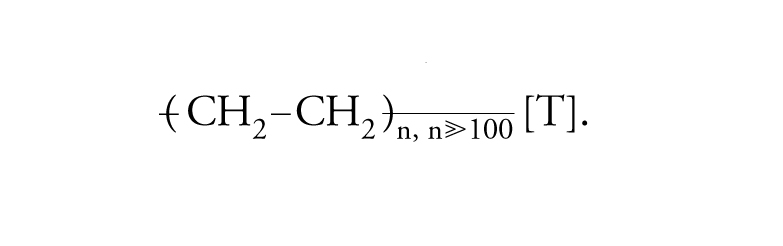
In ciascuna macromolecola di polietilene sono connesse a catena migliaia di unità monomeriche di composizione C2H4. Dal cloruro di vinile, CH2=CHCl, si producono le macromolecole che costituiscono il polivinilcloruro:
[I′] formula.
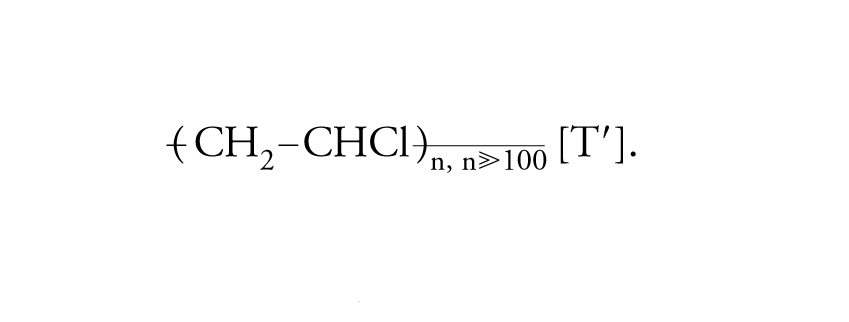
In ciascuna macromolecola del polivinilcloruro sono concatenate migliaia di unità monomeriche di composizione C2H3Cl. Dallo stirene, CH2=CH−C6H5, si ottengono le macromolecole che costituiscono il polistirene:
[I″] formula.
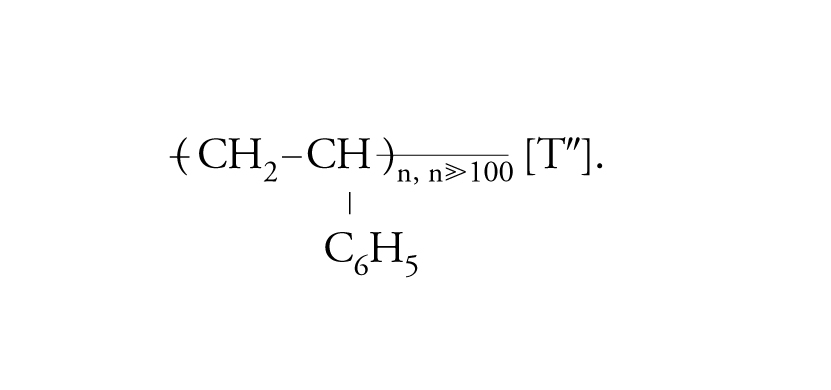
In ciascuna macromolecola del polistirene sono connesse a catena migliaia di unità monomeriche di composizione C8H8.
Polimerizzazione a stadi (o per condensazione), cioè per combinazione delle molecole di monomero o dioligomeri tra di loro, quasi sempre con perdita di molecole semplici, in genere molecole d'acqua. Per esempio, si ha la formazione di polimeri lineari attraverso la condensazione di un diacido di formula genera-le HO−CO−R−CO−OH con una diammina H2N−R′−NH2 o con un dialcol (glicole) HO−R″−OH (dove R, R′, R″ sono radicali organici bifunzionali), secondo successive reazioni del tipo:
HO−CO−R−CO−OH + H2N−R′−NH2
↓
HO−CO−R−CO−NH−R′−NH2 + H2O
oppure del tipo:
HO−CO−R−CO−OH + HO−R″−OH
↓
HO−CO−R−CO−O−R″−OH + H2O.
I prodotti di queste due reazioni, a loro volta, reagiscono con altre molecole di diacido, diammina o dialcol, causando la graduale crescita della catena. I polimeri così formati sono detti poliammidi e poliesteri. Le loro unità monomeriche sono rispettivamente:
−(CO−R−CO−NH−R′−NH−)n, n≥100
−(CO−R−CO−O−R″−O−)m, m≥100.
Così, il nylon è una poliammide di formula:
−[CO−(CH2)4−CO−NH−(CH2)6−NH−]n, n≥100
ottenuta a partire da acido adipico ed esametilendiammina.
Il terilene è un poliestere di formula:
−[CO−C6H4−CO−O−(CH2)2−O−]m, m≥100
ottenuto a partire dall'acido tereftalico e dal glicole etilenico.
Per quanto riguarda i polimeri di addizione sopra citati, la produzione industriale di polimeri del cloruro di vinile o PVC (polivinilcloruro) così come quella di polimeri dello stirene o PS (polistirene) incomincia agli inizi degli anni Trenta e raggiunge una dimensione significativa sia in Germania sia negli USA prima dello scoppio della Seconda guerra mondiale. Già in quegli anni, ne furono trovate molte applicazioni, nuove o sostitutive di altri materiali.
Polimeri del cloruro di vinile e polimeri dello stirene erano peraltro già noti da molti anni, anche se mal caratterizzati e con conoscenze assolutamente inadeguate su come utilizzarli. Un brevetto sul PVC fu dato in Germania a Fritz Klatte nel 1913, ma esso non venne mai sfruttato. La IG Farbenindustrie riprese a studiare il PVC a partire dal 1927.
Per quanto riguarda lo stirene, che è un liquido, era stato osservato e annotato che poteva trasformarsi in un solido gelatinoso, rigido a temperatura ambiente (che è poi il corrispondente polimero). La solidificazione avveniva (in apparenza) spontaneamente, con molta facilità, anche in assenza di altre sostanze aggiunte (iniziatori aggiunti), per effetto di impurezze presenti o di radiazioni assorbite.
Ancora per quanto riguarda i polimeri di addizione sopra citati, negli anni Trenta fu scoperto dai ricercatori della ICI (Imperial Chemical Industries) Reginald Gibson ed Eric Fawcett il metodo per ottenere il polietilene a bassa densità o LDPE (Low density polyethylene), e ne fu intrapreso lo sviluppo. Nel 1939 in Gran Bretagna la ICI aveva un impianto capace di produrre 100 t di materiale all'anno. Durante la guerra, il polietilene fu usato come isolante elettrico per i radar e le potenze dell'Asse non ne disponevano. Dopo la guerra, negli anni Cinquanta, l'LDPE, che si può ottenere con il processo ICI, è stato utilizzato come isolante in cavi elettrici di potenza, nella radio e nella televisione. Nel 2000, la produzione statunitense di politene, preparato sia ad alta pressione, come nel processo ICI, sia a bassa pressione, con nuovi processi messi a punto a partire dagli anni Cinquanta, è stata pari a circa 13 milioni di t, mentre la produzione mondiale nel 2001 era all'incirca 53 milioni di t.
Un'ulteriore scoperta (come vedremo per il caso del polietilene, a partire da una osservazione casuale, ma rilevata ed esaminata accuratamente), fu quella di Roy Plunkett nel 1938 sulla polimerizzabilità radicalica del politetrafluoroetilene (CF2=CF2) (in prova come gas per la refrigerazione). Il polimero risultante, commercializzato poi dalla Du Pont con il nome di teflon, è usato in numerosissime applicazioni domestiche sofisticate (dall'isolante elettrico, alle padelle antiaderenti, alle scarpe che traspirano). Per quanto riguarda i polimeri di condensazione ‒ nylon e terilene ‒ entrambi furono brevettati negli anni Trenta.
Grazie a ricerche condotte tra il 1927 e il 1935, Carothers riuscì a produrre per la Du Pont un polimero, il già citato nylon, che può essere filato in fibre di grande resistenza e di eccellenti prestazioni. Nel 1938 fu costruito il primo impianto di produzione 'da carbone, aria e acqua', come apparve nella pubblicità fattane all'epoca. Grande impressione fecero le calze di nylon: messe in vendita negli USA il 15 maggio 1940, una folla di donne ne comprò 4 milioni di paia in poche ore! Per ottenere una larga disponibilità di nylon per il consumo, però, fu necessario attendere alcuni anni dopo la fine della Seconda guerra mondiale, durante la quale invece le fibre di nylon vennero usate per la costruzione dei paracadute.
Anche lo sviluppo e la produzione di fibre di terilene, brevettate nel 1939, dovettero attendere la fine della guerra e gli anni Cinquanta. Nel 2000, la produzione statunitense di fibre di terilene (attualmente più spesso indicato con il nome derivato dalla sua struttura chimica, cioè polietilentereftalato, sigla PET) e di nylon (quasi 3 milioni di t) ha sorpassato di gran lunga quella delle fibre artificiali (meno di 200 mila t). Inoltre, PET e nylon sono prodotti e usati come materie plastiche di qualità per usi speciali, oltre che per la produzione di fibre.
È importante notare, infine, che tra la fine degli anni Quaranta e l'inizio degli anni Cinquanta avviene, per le materie prime della chimica industriale organica, una netta transizione dal carbone e suoi derivati (idrocarburi aromatici) al petrolio e agli idrocarburi alifatici. Un esempio è quello del cloruro di vinile, anche se indicazioni e ragionamenti analoghi potrebbero essere fatti per altri monomeri.
Negli anni Trenta, l'ottenimento del cloruro di vinile era possibile per via sintetica a partire dal carbone, materia prima prevalente all'epoca, per la realizzazione dei prodotti chimici organici industriali, per esempio dei coloranti.
Dopo la Seconda guerra mondiale, a partire dagli anni Cinquanta, con lo svilupparsi di nuove tecnologie e l'incremento dei flussi commerciali, la materia prima di partenza diventa il petrolio. Per la maggior parte dei prodotti organici di base, ivi compresi i monomeri, si passa da una chimica organica basata prevalentemente sul carbone a una basata prevalentemente sul petrolio; dalla chimica aromatica (degli idrocarburi aromatici, come benzene e derivati) alla chimica alifatica (degli idrocarburi alifatici, come, tra gli altri, etano, C2H6, etene o etilene, C2H4, propano, C3H8, propene o propilene, C3H6).
Nel caso del cloruro di vinile, esso veniva ottenuto, prima della Seconda guerra mondiale, attraverso il percorso sintetico di seguito descritto:
carbone (C) + calcare (CaCO3) (in forno elettrico)
→ carburo di calcio (CaC2)
carburo di calcio (CaC2) + H2O → acetilene (C2H2)
acetilene (C2H2)+HCl → cloruro di vinile (C2H3Cl).
Dopo la Seconda guerra mondiale, partendo da etilene, cloro e ossigeno, si è potuto ottenere direttamente per via della ossiclorurazione, e più economicamente, il cloruro di vinile, che, come si è detto, è il monomero di partenza del PVC.
6. La scoperta dei polimeri idrocarburici alifatici e della stereoregolarità
Agli inizi degli anni Cinquanta, come risultato di un intenso lavoro di ricerca scientifica, accompagnato da importanti realizzazioni industriali, la chimica delle macromolecole poggiava ormai su solide basi. Anche l'industria dei polimeri era in pieno sviluppo e produceva fibre (nylon, poliestere), gomme (gomma butile, gomma stirene-butadiene), varie resine termoplastiche (polietilene o politene, polivinilcloruro) e termoindurenti (per es., la bachelite).
I metodi di preparazione di cui allora disponeva la chimica macromolecolare, tuttavia, non erano in grado di fornire, partendo dai più comuni monomeri, come quelli vinilici e diolefinici, macromolecole a struttura ordinata. Dalla maggior parte di questi monomeri si ottenevano, infatti, polimeri amorfi o a bassissima cristallinità. Si era ben lontani dall'ordine strutturale dei polimeri naturali, tutti altamente cristallini.
Un decisivo progresso in questo campo fu raggiunto soltanto negli anni Cinquanta, attraverso le scoperte di Karl Waldemar Ziegler e Natta sull'ottenimento a bassa pressione di polimeri altamente cristallini dell'etilene (1953) e del propilene (1954), due olefine che si possono produrre dal petrolio, in maniera molto economica e in grandissime quantità. Queste scoperte costituiscono una tappa fondamentale nella storia della chimica macromolecolare, come illustreremo con qualche maggior dettaglio.
Abbiamo visto che alla fine degli anni Trenta la ICI in Gran Bretagna era stata in grado di produrre per la prima volta industrialmente polimeri dall'etilene. La scoperta era stata fatta casualmente, come spesso capita, da Fawcett e Gibson nel 1933, assoggettando ad altissime pressioni (oltre 1000 bar) e con intenti del tutto diversi da quelli di ottenere un polimero, una miscela di etilene e benzaldeide, contaminata, ma non di proposito, con ossigeno.
La riproducibilità e la stabilità della reazione di polimerizzazione fu ottenuta solo nel 1935, quando ci si rese conto della irrilevanza del sistema della benzaldeide e dell'importanza, invece, della contaminazione con ossigeno, che dava luogo a perossidi che si decomponevano a caldo per dare radicali liberi, essenziali iniziatori delle catene (indicati con [I]). Il polimero ha la costituzione ideale limite indicata in precedenza per il politene:
[I] formula
nella quale migliaia di unità monomeriche, di composizione C2H4, sono concatenate tra loro.
Oltre a queste unità monomeriche, nelle macromolecole del politene preparato ad alta pressione con iniziatori radicalici è presente un piccolo numero di ramificazioni lunghe e un certo numero di ramificazioni corte, del tipo:
−(CH−)
∣
CH2−CH2−CH2−CH2−CH3
che traggono origine da due unità etileniche.
Il polimero che si ottiene è parzialmente cristallino, e la sua struttura cristallina ‒ la prima a essere determinata per un polimero sintetico ‒ è stata descritta da Charles W. Bunn nel 1939. Durante la Seconda guerra mondiale, la disponibilità di politene permise alla Gran Bretagna e ai suoi alleati di progettare, produrre e installare i radar sulle navi e sugli aerei − il che contribuì significativamente al conseguimento della vittoria.
Agli inizi degli anni Cinquanta, Ziegler, direttore del Max-Planck-Institut für Kohlenforschung a Mülheim an der Ruhr (Germania), mise a punto con Hans Georg Gellert un'interessante reazione di crescita di catene etileniche su alluminiotrietile, Al(CH2CH3)3, che chiamò Aufbaureaktion (in inglese growth reaction) ossia reazione di crescita.
L'alluminiotrietile, alla pressione di circa 100 bar e alla temperatura di circa 100 °C, addiziona, sia pure con un processo lento, molecole di etilene sui legami metallo-carbonio, fino a che, dopo un certo nume-ro di addizioni, si staccano molecole di alchene C2H5−(−CH2−CH2)n−CH=CH2, con n compreso tra 0 e 100 (al massimo). La reazione non porta a veri e propri polimeri, ma a prodotti che si indicano come oligomeri (polimeri con catene molto corte, dal greco oligos, poco, e méros, parte; analogamente si può parlare di dimeri, trimeri, tetrameri, ecc.). Una caratteristica molto significativa della reazione è che le catene che si formano sono tutte assolutamente lineari, senza ramificazioni (che invece capitano, almeno in parte, per i polimeri dell'etilene nel processo ad alta pressione dell'ICI).
Un'osservazione interessante, indicativa di analogie e differenze, e dalla quale trassero indicazioni diverse Ziegler da una parte e Natta dall'altra, fu la seguente: partendo da alluminiotripropile Al(CH2CH2CH3)3 e propene, la reazione di addizione si arresta quasi quantitativamente a un solo dimero ben definito, il 2-metilpentene-1.
(CH2=C−CH2−CH2−CH3)
∣
CH3
In una conferenza di Ziegler, ascoltata a Francoforte nel 1952, Natta fu colpito dal racconto della straordinaria specificità di questa reazione di dimerizzazione del propene, come poi ha ricordato nella conferenza tenuta a Stoccolma all'atto del conferimento del premio Nobel nel 1963.
Nella primavera del 1953 nei laboratori di Ziegler fu notato che la Aufbaureaktion, che era sempre andata bene in centinaia di esperimenti, talvolta veniva meno dando luogo, invece, quantitativamente, al solo 1-butene (CH2=CH−CH2−CH3). Dopo una frenetica, rapida ricerca, Ziegler ed Erhard Holzkamp trovarono che piccole quantità di sali di nichel, che rimanevano nelle autoclavi di acciaio inossidabile dopo averle ripulite con acido nitrico, agivano come catalizzatori, trasformando la Aufbaureaktion in una reazione di dimerizzazione, come quella del propene. Questo fenomeno fu chiamato effetto nichel e diede inizio a ulteriori studi che portarono rapidamente alla scoperta dei catalizzatori a base di zirconio e titanio, due elementi di transizione del Gruppo 4 della tavola periodica degli elementi, che danno luogo alla sintesi di alti polimeri dell'etilene. Se un metallo, il nichel, era capace di influenzare così profondamente la reazione di crescita sugli alluminioalchili, che potevano fare altri metalli? Secondo Ziegler, la cosa ovvia da fare era una esplorazione sistematica della tavola periodica, provando volta per volta ciascun elemento metallico. Partendo così dal più piccolo dei polimeri dell'etilene, anzi, dal più piccolo oligomero, il butene, si pervenne rapidamente a imparare a fare il più alto polimero, il vero polietilene lineare.
Anziché sotto altissima pressione, come si doveva fare per ottenere il politene ICI, o sotto alta pressione, come si doveva fare per ottenere soltanto degli oligomeri nella Aufbaureaktion con alluminiotrietile, era possibile condurre la reazione di polimerizzazione dell'etilene in vitro, a pressione atmosferica e senza riscaldamento, con la sola aggiunta all'alluminiotrietile di tetracloruro di titanio (TiCl4).
Il polimero ottenuto rammolliva a temperature di 30 °C più alte del politene ICI, al disopra del punto di ebollizione dell'acqua, e poteva essere stampato in lastre molto più rigide e forti. In confronto, sempre con il politene ICI, classificato oggi come politene a bassa densità o LDPE (Low density polytene, nella normativa standard correntemente usata della ASTM-American Society for Testing and Materials Standards), il polimero preparato con catalizzatori al titanio era più cristallino e di densità maggiore, sicché è correntemente classificato come politene ad alta densità o HDPE (High density polytene).
Nel frattempo, attraverso un accordo stipulato tra Ziegler, Natta e la Montecatini, una grande società industriale italiana per la chimica, nei laboratori diretti da Natta presso l'Istituto di chimica industriale del Politecnico di Milano si era cominciato a studiare e caratterizzare dal punto di vista chimico-fisico e con la diffrazione dei raggi X sia gli oligomeri provenienti dalla Aufbaureaktion sia i polimeri ottenibili con i catalizzatori al titanio.
La scoperta della polimerizzazione del propilene a dare polimeri cristallini avvenne prima nei laboratori di Natta che non in quelli di Ziegler dove, una volta scoperto il modo di polimerizzare l'etilene, le ricerche furono orientate soprattutto nel trovare le combinazioni catalitiche migliori.
Nei laboratori dell'Istituto di chimica industriale del Politecnico di Milano, dove si svolgevano ricerche di chimica organica alifatica industriale era stata riscontrata la possibilità di polimerizzare l'etilene con l'aggiunta di tetracloruro di titanio all'alluminiotrietile.
L'obbiettivo era di provare la copolimerizzazione dell'etilene con il propilene (cioè l'addizione contemporanea, sulle stesse catene macromolecolari, e in rapporti regolabili, di etilene e propilene) con l'intento di preparare un materiale gommoso, anziché una materia plastica.
Nell'ambito di tale ricerca Paolo Chini caricò nel 1954, in un reattore riscaldato contenente solvente idrocarburico, alluminiotrietile e tetracloruro di titanio (catalizzatore e cocatalizzatore come si diceva allora), ottenendo una massa spugnosa verdastra, che fu esaminata ai raggi X dall'autore di questo articolo, che risultò essere inaspettatamente costituita da un materiale polimerico cristallino: il polipropilene isotattico.
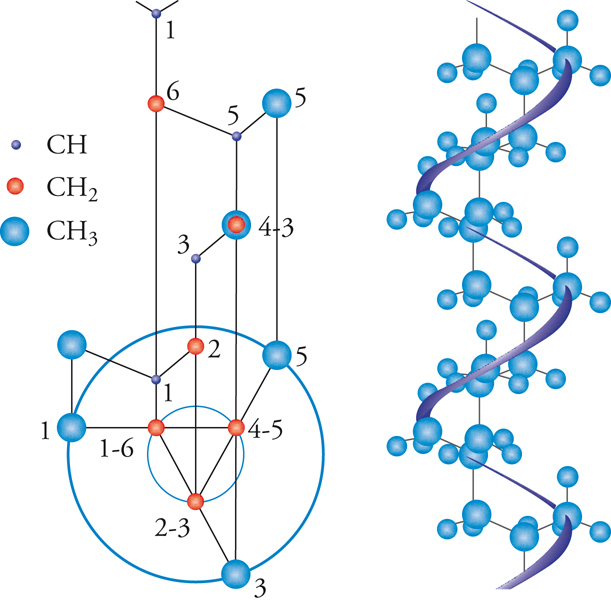
L'esame della struttura cristallina dei polimeri isotattici, in particolare del polipropilene, permise di stabilire che questi polimeri presentano tutti una conformazione elicoidale della catena (fig. 9). Strutture elicoidali di catena consentono in un polimero la ripetizione di unità monomeriche, che abbiano configurazioni successive identiche, in modo che esse assumano conformazioni equivalenti nei riguardi di un asse. Le ricerche ai raggi X furono perciò determinanti per attribuire una struttura isotattica ai polimeri cristallini da poco sintetizzati: dalla prima sintesi del polipropilene isotattico alla presentazione nel dicembre 1954 di due note all'Accademia dei Lincei, Una nuova classe di polimeri di α-olefine aventi eccezionale regolarità di struttura e Sulla struttura cristallina di un nuovo tipo di polipropilene passarono meno di nove mesi.
Alla scoperta dei polimeri isotattici di α-olefine ha fatto seguito la scoperta di numerosi altri polimeri stereoregolari. Nel caso dei polimeri dell'isoprene, per esempio, fu possibile ottenere e caratterizzare strutturalmente polimeri a concatenamento 1-4 con il doppio legame sempre cis o sempre trans, come nei due polimeri naturali già conosciuti, la gomma naturale e la guttaperca.
La catalisi stereospecifica di polimerizzazione, quindi, aveva permesso all'uomo di penetrare in un campo che, precedentemente, si riteneva un dominio assoluto della natura. I polimeri altamente cristallini ottenuti nel 1954 da Natta e dalla sua scuola a partire dalle principali α-olefine, che furono chiamati isotattici, sono caratterizzati dalla presenza di lunghe sequenze di unità monomeriche aventi la stessa configurazione.
Successivamente Natta e la sua scuola ottennero dal propilene polimeri stereoregolari di altro tipo, caratterizzati dalla presenza di lunghe sequenze di unità monomeriche aventi alternativamente configurazione opposta. Tali polimeri furono chiamati sindiotattici. Polimeri altamente regolari furono ottenuti anche dal butadiene e da altre diolefine. Questi risultati stimolarono in tutto il mondo ricerche sulla polimerizzazione stereospecifica, che hanno portato alla realizzazione di numerosi polimeri stereoregolari, ottenuti non solo da monomeri idrocarburici lineari, vinilici e diolefinici, ma anche da cicloolefine, aldeidi, epossidi e monomeri acrilici.
La scoperta della polimerizzazione stereospecifica ha aperto un periodo nuovo nella chimica macromolecolare, il cui interesse non è solo scientifico, ma anche pratico; l'industria delle materie plastiche, degli elastomeri e delle fibre ne è stata infatti profondamente influenzata.
Nel 1963 Ziegler e Natta hanno ricevuto insieme il premio Nobel per la chimica. Arne Fredga, nel discorso di presentazione per il premio, ha messo in evidenza come "la nostra epoca stia assistendo al graduale rimpiazzo di materiali tradizionali (quali vetro, porcellana, legno, metalli) con materiali sintetici, le materie plastiche, ottenibili per polimerizzazione (di molecole piccole).
Ziegler ha inventato un metodo interamente nuovo di polimerizzazione, in particolare dell'etilene, realizzando la crescita delle catene polimeriche (completamente lineari) per addizione su legami metallo-carbonio. Questa addizione catalitica è molto più dolce di quella nota precedentemente su radicali liberi, che può dare invece catene con ramificazioni o altre anomalie.
Se invece dell'etilene si polimerizza il propilene, questo potrebbe dar luogo a catene, con un gruppo laterale metilico −(CH3) ogni due atomi di carbonio; questo gruppo laterale potrebbe essere orientato a destra oppure a sinistra lungo la catena. Quando questi orientamenti sono distribuiti a caso, la catena ha una configurazione spaziale irregolare. Natta, tuttavia, ha trovato che certi tipi di catalizzatori Ziegler portano a macromolecole stereoregolari, cioè a macromolecole con una struttura spazialmente uniforme. In queste catene tutti i gruppi laterali puntano a destra, oppure tutti a sinistra, e le catene si dicono isotattiche. L'intorno molecolare dell'atomo di metallo, sul quale si addizionano le unità monomeriche, ha una forma tale da permettere una sola orientazione definita per i gruppi laterali".
Queste dichiarazioni di Fredga, e le dichiarazioni analoghe di Natta nel discorso d'investitura, si possono considerare profetiche rispetto a ritrovati scientifici e conclusioni certe ottenute molto più recentemente.
Nella motivazione di Fredga si legge ancora: "I polimeri isotattici mostrano caratteristiche molto interessanti. Mentre le catene idrocarburiche ordinarie hanno una forma a zig-zag, le catene isotattiche formano eliche, da cui i gruppi laterali spuntano in fuori. Questi polimeri danno origine a nuovi prodotti sintetici, come tessuti che sono leggeri e forti nello stesso tempo e funi che galleggiano sull'acqua, per citare solo due esempi.
La natura sintetizza molti polimeri stereoregolari, per esempio la cellulosa e la gomma. Si pensava finora che questo fosse un monopolio della natura, che opera con biocatalizzatori noti come enzimi. Ma adesso il prof. Natta ha spezzato questo monopolio".
La scoperta dei polimeri stereoregolari ha prodotto ‒ e sta ancora producendo ‒ importanti conseguenze nel campo della scienza pura e applicata.
Può essere interessante fare un rapido esame dei più recenti miglioramenti apportati nel campo della catalisi stereoselettiva. Alla fine degli anni Sessanta, partendo dai primi catalizzatori Ziegler-Natta a base di cloruro di titanio, vennero implementati nuovi catalizzatori per la polimerizzazione dell'etilene, nei quali il cloruro di titanio è supportato su una matrice, per esempio ossido o cloruro di magnesio solidi. Questi nuovi catalizzatori mostravano un'altissima attività nella polimerizzazione dell'etilene, con rese di 1000, anziché 10, chilogrammi di polimero per grammo di titanio. I nuovi catalizzatori, però, non erano soddisfacenti per la polimerizzazione del propilene, dove il controllo delle successioni di configurazioni relative meso o racemiche è assolutamente necessario.
Negli anni Settanta, le ricerche intese a progettare e trovare catalizzatori supportati ad altissima resa anche per il propilene hanno avuto successo. I nuovi sistemi catalitici danno luogo a rese così alte (migliaia di chilogrammi di polipropilene isotattico per grammo di titanio), che i processi di depurazione dal catalizzatore possono essere eliminati negli impianti di produzione; e anche il costo gravoso di estrazione della frazione amorfa (che accompagnava il polimero nei precedenti processi di produzione) può essere eliminato. Attualmente, molecole di dieteri appositamente disegnate condizionano i catalizzatori supportati ad attività e stereospecificità estremamente elevate usati nell'industria.
Gli sforzi di ricerca per una migliore comprensione del meccanismo d'azione dei catalizzatori Ziegler-Natta, della natura delle superfici dei catalizzatori solidi e dell'influenza di vari agenti chimici hanno dunque ben pagato, anche in termini di un processo industriale più semplice ed ecologicamente corretto. E la scoperta, negli anni Ottanta e in quelli successivi, di catalizzatori solubili, anziché solidi, ha aperto nuove prospettive per una polimerizzazione più regolabile nella costituzione e nella configurazione per i polimeri ottenibili, principalmente, ma non esclusivamente, dall'etene e dal propene.
7. Sviluppi attuali della scienza e delle applicazioni dei polimeri
Gli studi delle conformazioni macromolecolari, della struttura cristallina e quindi del modo di impacchettamento delle catene e della morfologia dei materiali polimerici, hanno ricevuto un notevole impulso dalla scoperta dei polimeri stereoregolari e dei numerosi altri che sono stati e vengono continuamente sintetizzati. Per quanto riguarda le conformazioni macromolecolari, la prima conformazione a elica determinata per un polimero sintetico è stata quella del polipropilene isotattico che, come abbiamo ricordato in precedenza, fu pubblicata nel 1954. A essa sono seguite numerose altre. Quasi contemporaneamente, erano state proposte strutture a elica per importanti macromolecole naturali, da Linus C. Pauling per i poli-α-amminoacidi (premio Nobel per la chimica nel 1954), da Francis H.C. Crick e James D. Watson per il DNA (premi Nobel per la medicina con Maurice H.F. Wilkins nel 1962).
Gli studi sulla struttura cristallina e sul modo di impacchettamento delle catene nei microcristalli dei polimeri sono stati portati avanti inizialmente dalla Scuola di Natta e approfonditi successivamente da questa e da numerosi altri ricercatori. Gli studi di morfologia (cioè del modo di organizzarsi dei microcristalli nei solidi polimerici) sono stati portati avanti dalla Scuola di fisica dei polimeri fondata da Andrew Keller. A lui è dovuta la scoperta che il polietilene lineare può cristallizzare da soluzione in cristalli singoli di forma lamellare, di spessore nanometrico. In tali cristalli singoli, le catene polimeriche di ogni macromolecola possono ripiegarsi più e più volte.
La chimica fisica dei polimeri e la termodinamica statistica delle macromolecole hanno avuto un grande cultore in Paul J. Flory, che ha ricevuto il premio Nobel per la chimica nel 1974 e presso il quale si sono formati molti ricercatori accademici e industriali negli USA e nel mondo.
Le macromolecole in soluzione o allo stato fuso sono rappresentabili come un gomitolo, mentre allo stato solido si possono organizzare in microcristalli contenenti catene parallele tra loro, se il polimero ha una struttura chimica che lo rende capace di cristallizzare. In tal caso risulta che, allo stato solido, le catene macromolecolari possono appartenere a zone cristalline oppure a zone amorfe, e il polimero si dice semicristallino. I microcristalli, a loro volta, sono collegati da catene macromolecolari (tie-chains), che sono molto importanti per conferire ai materiali polimerici semicristallini proprietà meccaniche di tenacità e di resistenza a rottura.
Gli studi chimico-fisici, strutturali e morfologici, sono risultati di grande importanza nella caratterizzazione e nello studio delle relazioni tra proprietà e struttura chimica, relazioni che sono appunto mediate dalla struttura fisica e dalla sua evoluzione temporale (che ricomprende la lavorazione e il comportamento nell'uso). L'applicazione di un materiale polimerico per un uso specifico, infatti, è legata, e spesso in modo assai complesso, alla struttura fisica e all'evoluzione temporale di tale struttura.
Si può parlare di polimeri tailor-made, intendendo riferirsi a costituzioni chimiche mirate a una utilizzazione precisa. A tale proposito si possono ricordare le fibre aramidiche, poliammidi aromatiche usate nei corsetti antiproiettile per la loro leggerezza, unita a una resistenza emula dell'acciaio; un esempio ne è il Kevlar della Du Pont, messo a punto agli inizi degli anni Settanta, o, ancora, il polivinilidenfluoruro, −(CH2CF2−)n, che è usato quando occorra un isolante elettrico dotato anche di grande resistenza al calore e al fuoco, e che è il materiale piezoelettrico utilizzato correntemente per tradurre segnali elettrici in suoni negli altoparlanti, e i policarbonati di cui si utilizza la perfetta trasparenza e la rigidità alle temperature di uso: le applicazioni attuali, in crescita continua (500 mila t all'anno nel mondo nel 2000), vanno dalla fabbricazione di dischi per la raccolta informatica di quantità sempre più grandi di dati, testi, suoni e immagini, alla fabbricazione di grandi coperture architettoniche trasparenti.
Si può ricordare, infine, sempre a proposito di polimeri tailor-made, che il premio Nobel per la chimica 2000 è stato conferito ad Alan J. Heeger, Alan G. MacDiarmid e Hideki Shirakawa per aver dimostrato che il poliacetilene lineare, (−CH=CH−)n (un polimero di lucentezza metallica, preparato per la prima volta da Natta e collaboratori con catalizzatori metallorganici), e altri polimeri contenenti doppi legami coniugati possono essere portati ad avere conducibilità metallica, se opportunamente drogati. Non solo, dunque, le materie plastiche sono normalmente buoni o addirittura eccellenti isolanti elettrici, ma, sotto certe circostanze, possono comportarsi come un metallo.
Come detto da Flory nella sua conferenza Nobel, si può prevedere anche per il futuro che studi del tipo sopra riportato permetteranno di comprendere sempre meglio, in termini fondamentali, le marcate differenze in proprietà che distinguono la grande varietà di sostanze polimeriche, sia naturali sia sintetiche.
Lavorazione e destinazioni d'uso delle materie plastiche
La plasticità è la capacità di un materiale di cambiare la propria forma sotto l'azione di una forza relativamente piccola e di mantenere la forma raggiunta quando la forza viene rimossa. Perché un materiale dotato di plasticità abbia interesse commerciale, deve manifestare tale sua caratteristica durante la lavorazione (in modo da poter essere foggiato nella forma desiderata), ma deve perdere tale caratteristica nelle condizioni normali d'uso. L'uomo ha utilizzato la plasticità di alcuni materiali fin dagli albori della civiltà: egli imparò presto che un impasto di acqua e argilla può essere modellato, semplicemente sotto la pressione delle dita, nella forma di vasi e stoviglie e può mantenere la forma acquistata se posto in un forno, dove perde la plasticità per trasformarsi in un solido rigido.
Come affermato in precedenza, materie plastiche di natura polimerica erano note all'uomo fin dall'Antichità. Ne sono un esempio l'ambra fossile e la guttaperca, quest'ultima molto usata in Occidente nell'Ottocento e ancora nella prima metà del Novecento, ricavabile da alberi malesi della famiglia delle Sapotacee. Ma soltanto in tempi recenti si è pervenuti alla scoperta, alla produzione per sintesi e alla utilizzazione di una varietà enorme di tali materiali, il cui consumo è aumentato esponenzialmente negli ultimi anni.
A seconda del modo in cui può essere conferita una forma definita agli oggetti, le materie plastiche possono essere suddivise nelle due categorie di materie termoplastiche (la maggior parte prodotta) e materie termoindurenti.
Le materie termoplastiche sono dure e rigide a temperatura ambiente; rammolliscono e diventano plastiche (nelle parole del premio Nobel 1991 per la fisica Pierre-Gilles de Gennes, diventano materia soffice) a temperature più o meno elevate, a seconda del materiale considerato (fra i 150 °C e i 200 °C per le materie termoplastiche più usate). A tali temperature può essere loro conferita la forma desiderata. A temperatura ambiente, il materiale termoplastico diventa di nuovo rigido (a differenza del termoindurente, come la bakelite), ma se riscaldato di nuovo può cambiare un'altra volta forma.
I principali procedimenti che trasformano i polimeri (forniti normalmente dall'industria in polvere o granuli) nei prodotti finali, utilizzando calore e pressione, sono la calandratura, l'estrusione, il soffiaggio e lo stampaggio.
La calandratura fa passare il polimero tra cilindri riscaldati e consente di ottenere in continuo fogli dello spessore desiderato. L'estrusione, che è il procedimento più diffuso, consiste nella trasformazione in continuo di materiale plastico riscaldato e spinto da una vite senza fine, attraverso un ugello che dà al materiale la sagoma richiesta; per raffreddamento l'oggetto assume la sua forma stabile. Nel soffiaggio il polimero fuso viene sottoposto a soffiaggio con aria o vapore in modo da assumere la forma dello stampo in cui viene alimentato. Lo stampaggio è la tecnica che vede il polimero fuso alimentare uno stampo di cui, per compressione e raffreddamento, assume la forma desiderata. Tutti questi procedimenti hanno attraversato innovazioni tecnologiche continue che hanno affiancato i progressi della scienza dei polimeri negli ultimi decenni.
I materiali polimerici trovano applicazioni nei settori più disparati: nell'edilizia, nei trasporti, nelle comunicazioni, nell'agricoltura e nell' industria alimentare, negli imballaggi, nella medicina e salute dell'uomo, negli articoli sportivi e per il tempo libero.
Secondo i dati Federchimica-Assoplast, in Italia nel 2000 l'edilizia avrebbe assorbito oltre l'11% della produzione di materie plastiche (13,2% nel mondo). Impieghi tipici sono tubazioni, membrane per impermeabilizzazione di coperture, cavi elettrici e guaine per isolamento.
I trasporti assorbono nel mondo il 7% della produzione di materie plastiche, utilizzate in numerose componenti delle automobili. Molte utilizzazioni, come air bag e cinture di sicurezza, sono rese possibili proprio dalla plastica. Il contenuto medio di materie plastiche in un'auto europea è passato da circa 20 kg negli anni Sessanta (2% in peso) ai circa 105 kg del 2000 (in media circa il 10% del peso totale dell'auto).
Gli imballaggi assorbono nel mondo il 43,5% di tutta la produzione di materie plastiche (in Italia, oltre il 45%). In Europa, circa il 50% di tutto l'imballaggio alimentare è in plastica, e il 60% di questo tipo di confezioni pesa meno di 10 g. In 20 anni, con la disponibilità di più adatti materiali polimerici e il miglioramento delle tecnologie di lavorazione, gli imballaggi (packaging) si sono alleggeriti dell'80%, e le prestazioni sono migliorate.
Per quanto riguarda la salute, negli impieghi delle materie plastiche si va dagli imballaggi termoformati dei farmaci, ai numerosissimi oggetti e attrezzature impiegati in medicina e chirurgia: guanti sterili, siringhe per iniezioni, sacche per il trasporto di sangue e plasma, protesi chirurgiche.
Per quanto riguarda l'impiego di materie plastiche specifiche, un materiale che si sta facendo spazio nell'uso è il polipropilene di cui si sono prodotti nel 2000, oltre 20 milioni di t all'anno nel mondo. Così, tutte le siringhe con cui si fanno attualmente le iniezioni, tutte le sedie non di legno che vediamo nei bar o che usiamo nelle nostre case di campagna, tutti i contenitori delle batterie delle automobili, sono di questo polimero.
8. Lo sviluppo temporale dell'industria dei polimeri
All'inizio degli anni Duemila, una parte significativa della produzione industriale nel mondo riguarda i materiali polimerici, ripartita tra USA (per circa un quarto), Europa occidentale (per circa un quarto) e i restanti paesi, tra i quali un ruolo importante ha il Giappone.
La produzione industriale di polimeri nel mondo era molto limitata all'inizio della Seconda guerra mondiale ed è letteralmente esplosa nel periodo 1950-2000. Riteniamo utile dare un quadro indicativo dell'evoluzione della produzione industriale di polimeri attraverso i dati disponibili per gli USA. Faremo riferimento ai rapporti annuali pubblicati dal giornale dell'American Chemical Society, "Chemical and engineering news".
Nel rapporto annuale sulla produzione chimica pubblicato nel 2002, la produzione di polimeri è riportata nelle categorie delle materie plastiche, della gomma sintetica e delle fibre sintetiche. Per il 2000, la produzione USA di resine termoplastiche è stata di oltre 32 milioni di t. La frazione di materiali polimerici che costituiscono le resine termoindurenti (tipo la bakelite, che ne è stata l'antesignana) si avvicina ormai solo al 10% della produzione totale di resine termoplastiche.
È da notare che nel 1960 la produzione industriale USA di materie plastiche è riportata da "Chemical and engineering news" in 2,7 milioni di t, con il commento di aver raggiunto completa maturità. Da questo dato, e da quello sopra riportato per il 2000, si evince invece che la produzione di materie plastiche si è più che decuplicata (precisamente, è aumentata di un fattore 12) dal 1960 al 2000, essendo stato l'aumento medio, a partire dal 1960, anno della presunta maturità, del 6,4% all'anno nell'arco di ben 40 anni.
Le resine termoplastiche sono costituite principalmente da polietilene a bassa e alta densità, da polipropilene, da polistirene e da polivinilcloruro. I dati sulla produzione USA di questi polimeri per gli anni 1960-2000 evidenziano una rapida crescita, praticamente da zero, nel cinquantennio scorso.
La crescita più rapida è stata quella del polipropilene che ha raggiunto e superato alla fine del secolo scorso una produzione di 20 milioni di t in tutto il mondo (per un consumo di più di 3 kg all'anno per abitante della Terra).
Anche il polietilene ha avuto una rapida crescita. Con i catalizzatori di Ziegler si può preparare il polietilene ad alta densità, più duro e con punto di fusione più alto del polietilene a bassa densità. Come si è già accennato, quest'ultimo nel processo originale ICI si preparava (e tuttora, in parte, si prepara) ad alta pressione, mentre si può ottenere per lo più, per oltre la metà, a bassa pressione con catalizzatori metallorganici, attraverso la copolimerizzazione dell'etilene con altre olefine (questo materiale è talvolta denominato LLDPE, Linear low density polyethylene). La produzione USA nel 2000 di LDPE è di 3436 t, di LLDPE è di 3606 t, per un totale di 7042 t.
Sempre per il 2000, la produzione di gomma sintetica è stata di circa 2,5 milioni t, quella di fibre sintetiche di quasi 5 milioni t, mentre quella di fibre artificiali, acetato di cellulosa e rayon, si è ormai ridotta a 158 mila t.
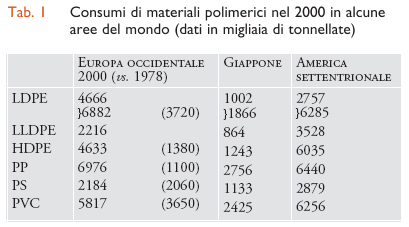
Dati analoghi sui consumi di materie plastiche in Europa e in Giappone, in confronto con l'America settentrionale, sono riportati per il 2000 nella tab.1.
Bibliografia
Baum 1998: Baum, Rudy M., Chemistry's golden age, "Chemical and engineering news", 76/2, 1998, pp. 143-151.
Brandrup, Immergut 1989: Brandrup, Johannes - Immergut, Edmund H., Polymer handbook, 3. ed., New York-Chichester, Wiley, 1989 (1. ed.: New York, Interscience, 1966).
Bunn 1939: Bunn, Charles W., The crystal structure of long-chain normal paraffin hydrocarbons. The shape of the -CH2 group, "Transactions of the Faraday Society", 35, 1939, pp. 428-491.
Chenier 1992: Chenier, Philip J., Survey of industrial chemistry, 2. ed., New York, VCH, 1992, pp. 271-370 (1. ed.: New York, Wiley, 1986).
Corradini 1982: Corradini, Paolo, Ruolo della scoperta e studio dei polimeri stereoregolari nella chimica macromolecolare, in: Attuali prospettive nella scienza dei polimeri, Roma, Accademia Nazionale dei Lincei, 1982, pp. 11-38.
Corradini 1986: Corradini, Paolo, Ordine strutturale nei polimeri, "Rendiconti dell'Accademia Nazionale dei Lincei", 80, 1986, pp. 261-279.
Corradini 1995: Corradini, Paolo, The impact of the discovery of stereoregular polymers in macromolecular science, "Macromolecular symposia", 89, 1995, pp. 1-11.
Corradini 1998: Corradini, Paolo, Regio- e stereoselettività nelle polimerizzazioni con catalizzatori Ziegler-Natta, in: Enzimi e catalizzatori chimici, Roma, Accademia Nazionale dei Lincei, 1998, pp. 43-76.
Corradini, Nicolais 1989: Corradini, Paolo - Nicolais, Gino, Materiali polimerici, Roma, Istituto della Enciclopedia Italiana, 1989, pp. 673-688.
Corradini, Paiaro 1970: Corradini, Paolo - Paiaro, Gastone, Polimeri. Materie plastiche, fibre e gomme, in: Il secondo libro di chimica per gli istituti tecnici industriali. Corso di chimica organica e industriale, Firenze, Giunti Bemporad Marzocco, 1970, pp. 133-166.
Corradini, Porri 1981: Corradini, Paolo - Porri, Lido, Polimeri, in: Enciclopedia del Novecento, Roma, Istituto della Enciclopedia Italiana, V, 1981, pp. 423-451.
Flory 1953: Flory, Paul J., Principles of polymer chemistry, Ithaca (N.Y.), Cornell University Press, 1953.
Furukawa 1998: Furukawa, Yasu, Inventing polymer science. Staudinger, Carothers, and the emergence of macromolecular chemistry, Philadelphia, University of Pennsylvania Press, 1998.
MacMillan 1979: MacMillan, Frank M., The chain straighteners. Fruitful innovation: the discovery of linear and stereoregular synthetic polymers, London, Macmillan, 1979.
Metanomski 1991: Compendium of macromolecular nomenclature, edited by W. Val Metanomski, Oxford, Blackwell Scientific, 1991.
Moore 1996: Polypropylene handbook: polymerization, characterization, properties, applications, edited by Edward P. Moore jr, Munich-New York, Hanser, 1996.
Morawetz 1985: Morawetz, Herbert, Polymers. The origins and growth of a science, New York, Wiley, 1985.
Reisch 1998: Reisch, Marc S., From coal tar to crafting a wealth of diversity, "Chemical and engineering news", 76, 1998, p. 90.
Seymour 1982: Seymour, Raymond B., History of polymer science and technology, New York-Basel, Dekker, 1982.
Seymour, Cheng 1986: Seymour, Raymond B. - Cheng, Tai, History of polyolefins: the world's most widely used polymers, Dordrecht-Boston, Reidel, 1986.
Ziegler 1960: Ziegler, Karl W., Organoaluminium compounds, in: Organometallic chemistry, edited by H. Zeiss, New York, Reinhold, 1960, pp. 294-296.
Rapporti sulle produzioni chimiche negli Stati Uniti e nel mondo sono pubblicati annualmente (Facts and figures for the chemical industry) nella rivista della American Chemical Society, "Chemical and engineering news" (per es. nel n. 25 del 24 giugno 2002). Sempre nella stessa rivista compaiono periodicamente rassegne storiche, per es. nel n. 2 del 12 gennaio 1998.