
Microarray
Microarray
È nozione acquisita che migliaia di geni e i prodotti da essi codificati (RNA e proteine) partecipano in maniera complessa e coordinata ai meccanismi che sono alla base della vita di ogni organismo. I biologi, pur consapevoli di questa enorme complessità degli organismi viventi, non disponevano, fino a poco tempo fa, di tecnologie di indagine adeguate per affrontarne lo studio. La biologia molecolare tradizionale operava, infatti, sulla base del criterio ‘un esperimento, un gene’, adatto per chiarire singoli processi biologici, ma assolutamente insufficiente per affrontare lo studio dello sviluppo e del funzionamento di un organismo nel suo insieme. La tecnologia dei microarray di DNA, sviluppatasi negli ultimi anni, ha suscitato un enorme interesse e promette di poter rispondere a questa sfida. Si tratta, infatti, di una tecnica che permette di esaminare in parallelo, in maniera veloce ed economica, l’intero genoma di un organismo o la totalità dei suoi prodotti su una singola lastrina di vetro o di silicio, un chip.
La tecnologia dei microarray di DNA per lo studio degli acidi nucleici si basa sulla proprietà di questi ultimi di ibridare, cioè di riassociarsi con la propria sequenza complementare secondo le regole di appaiamento delle basi scoperte da Watson e Crick. Questa caratteristica degli acidi nucleici è alla base di molte delle loro proprietà biologiche nonché delle tecnologie tradizionali per studiare l’espressione genica, quali il Southern e il Northern blot. Grazie a questa proprietà, un frammento di acido nucleico può funzionare da sonda specifica per il riconoscimento della sua sequenza complementare, e permette quindi di identificarla anche in una miscela complessa. Il processo di ibridazione è estremamente selettivo, specifico e sensibile. Nelle tecniche tradizionali la sonda è costituita da più molecole identiche di acido nucleico marcate generalmente con un isotopo radioattivo. Le molecole della sonda si trovano in fase liquida e solo un tipo di sonda alla volta può essere ibridata alle sequenze bersaglio dei campioni in esame, che sono invece ancorate a un supporto. Una volta che l’ibridazione è avvenuta e le molecole di sonda in eccesso sono state eliminate, sarà possibile per mezzo di autoradiografia visualizzare e quantificare il segnale emesso dalla sonda ibridata, che sarà proporzionale alla quantità di sequenza specifica contenuta nei campioni in esame. Nel caso del Southern e del Northern blot, tecnologie ormai classiche in biologia molecolare, i campioni esaminati sono sottoposti a elettroforesi in gel di agarosio prima dell’ibridazione, e quindi trasferiti su un filtro utilizzato come supporto. In tal caso, dopo l’ibridazione la sonda evidenzierà non solo la quantità di sequenza bersaglio presente in ciascun campione ma anche il suo peso molecolare.
Le tecnologie basate sui microarray ribaltano la relazione fra sonde e sequenze bersaglio. In questo caso, su uno stesso supporto sono ancorate migliaia di sonde diverse, non marcate, mentre il campione da esaminare si trova in fase liquida ed è marcato, in genere con un fluorocromo. L’ibridazione avverrà in parallelo, contemporaneamente tra tutte le sonde e le rispettive sequenze bersaglio contenute nel campione. Dopo l’ibridazione e l’eliminazione del campione che non ha reagito perché in eccesso, si visualizzerà e quantificherà il segnale fluorescente rimasto legato a ciascuna sonda: esso sarà proporzionale alla quantità di sequenza complementare riconosciuta dalla specifica sonda nel campione in esame.
Strutturalmente, i microarray sono costituiti da un supporto solido su cui sono disposte ordinatamente un numero elevato di sonde specifiche di DNA in maniera da formare una matrice di punti regolare. Ogni punto della matrice ha dimensioni tipicamente inferiori a 200 micron ed è costituito da molte copie della stessa sequenza di DNA. Esso rappresenta l’unità minima del microarray ed è chiamato feature. I microarray si classificano per il numero di features presenti sulla loro superficie, una sorta di misura della loro complessità e capacità di risoluzione. Attualmente sono in commercio microarray che dispongono di più di un milione di features. Ogni feature è costituita da più copie uguali di sequenze sonda (probe sequences), che ibrideranno con le sequenze bersaglio (target sequences) complementari marcate contenute nei campioni in esame. Dai microarray è quindi possibile ottenere un’enorme mole di dati, la cui gestione richiede lo sviluppo di adeguati strumenti informatici, dei quali va sottolineata l’importanza per l’evoluzione futura della ricerca in questo ambito.
Tecnologie costruttive
Attualmente dal punto di vista costruttivo i microarray possono essere divisi in due classi a seconda del tipo di acido nucleico usato per la composizione delle sonde: i microarray con sonde di cDNA e quelli con sonde di oligonucleotidi.
Inizialmente i microarray sono stati costruiti utilizzando migliaia di sonde, ciascuna costituita dal cDNA di un gene specifico, depositate ordinatamente a formare una matrice sulla superficie di un supporto adeguato. Le sonde in genere sono ottenute mediante amplificazione con PCR da plasmidi accuratamente caratterizzati derivati da genoteche di sequenze EST clonate. Queste genoteche sono preparate da cDNA derivati per retrotrascrizione da RNA messaggeri e quindi sono rappresentative delle sequenze geniche espresse nei vari tessuti di origine, da cui il nome EST (Expression sequence tag). Le sonde purificate sono quindi stampate per contatto da un robot munito di pennini molto sottili su un vetrino standard da microscopia rivestito di polilisina. Per ogni punto della matrice il pennino deposita circa 0,6 μL, formando una macchiolina che contiene da 1 a 10 ng di DNA e ha un diametro di circa 100 μm.
Questa tecnologia, ancora oggi molto usata, presenta però notevoli problemi. Obbliga a conservare e gestire grandi genoteche di cDNA, con tutte le difficoltà legate alla loro manutenzione, annotazione e controllo delle identità. Inoltre, le diverse sequenze di cDNA usate come sonda, spesso differiscono molto fra di loro per lunghezza, contenuto di guanine e citosine e presenza di sequenze ripetute. A sua volta, questa variabilità si riflette in ampie differenze delle proprietà di amplificazione tramite PCR e in scarsa omogeneità di ibridazione, aumentando notevolmente gli errori del sistema.
Per ovviare a questi problemi, in seguito anche alla disponibilità della sequenza del genoma di molti organismi, negli ultimi anni si sono diffusi microarray costruiti con lunghi oligonucleotidi (50÷70 nucleotidi) singola elica, sintetizzati artificialmente e raccolti in collezioni disponibili commercialmente. Questi oligonucleotidi sono progettati da programmi software che interrogano automaticamente le basi di dati genomiche e ne estraggono sequenze sonda rappresentative, uniche e specifiche per i singoli geni, ottimizzandone le proprietà di ibridazione. La gestione di queste raccolte è molto più semplice se paragonata a quella delle genoteche di plasmidi e in genere l’annotazione è molto più accurata.
Negli ultimi anni hanno acquistato sempre più importanza piattaforme tecnologiche gestite da grandi ditte che forniscono microarray ad altissima densità, con più di 1 milione di feature per array, pronti all’uso, che coprono tutto il genoma delle specie animali e vegetali più utilizzate nella ricerca e nell’industria.
Tra le diverse piattaforme disponibili, una delle più utilizzate (attualmente prodotta dall’azienda californiana Affymetrix) sfrutta una tecnologia in parte mutuata da quella usata per costruire i chip semiconduttori elettronici, basata sulla fotolitografia e sulla chimica in fase solida, che permette di sintetizzare in situ milioni di oligonucleotidi su un piccolo chip di quarzo. Il wafer di quarzo, che costituirà il supporto di una serie di microarray identici, è ricoperto da uno strato protettore fotosensibile che impedisce il legame chimico dei nucleotidi monomeri che serviranno a sintetizzare le sequenze degli oligonucleotidi. Il supporto, quindi, è esposto attraverso una prima maschera fotolitografica alla luce che illuminerà solo le zone destinate, per esempio, a legare il nucleotide adenina. L’esposizione alla luce renderà solubile lo strato fotosensibile e deproteggerà il substrato. Il wafer è poi esposto a una soluzione contenente il nucleotide adenina, che potrà legarsi solo alle zone del supporto deprotette, dove costituirà il primo nucleotide della sonda desiderata in quel punto. L’operazione quindi è ripetuta in successione con gli altri 3 nucleotidi, guanina, timidina e citosina, usando ogni volta una nuova apposita maschera che permetterà di deproteggere solo le zone destinate a ciascun nucleotide. Ovviamente anche le estremità libere dei nucleotidi che si sono legati sono protette chimicamente da un gruppo fotosensibile, cosicché a ogni ciclo se ne può legare soltanto uno e il processo può continuare solo quando questo gruppo sarà rimosso per esposizione alla luce. In tal modo, per cicli successivi di deprotezione e sintesi, un nucleotide alla volta, milioni di sonde crescono con la sequenza desiderata fino a raggiungere la lunghezza prefissata, in genere di 25 nucleotidi. Infine, il wafer di quarzo su cui sono state sintetizzate tutte le sonde, è tagliato nei singoli chip costituenti.
A questa avanzata tecnologia costruttiva è stata affiancata una tecnologia bioinformatica altrettanto avanzata. Nel caso dei microarray destinati all’analisi dei profili di espressione genica, ogni trascritto è rappresentato sul microarray da un set di almeno 11 coppie diverse di feature, ciascuna formata da sonde complementari a una diversa zona del trascritto. Nell’ambito di ciascuna coppia di feature, una è costituita da oligonucleotidi perfettamente complementari alla sequenza di riferimento (sonda PM, perfect match), mentre l’altra è costituita da oligonucleotidi che presentano almeno un errore di appaiamento con la sequenza di riferimento (sonda MM, mismatch). Complessivamente, un set completo di sonde è costituito da due tipi di sequenze, uno PM e l’altro MM, per un totale di 250÷300 coppie di basi ciascuno. Dato che la presenza di una singola base non corretta è capace di destabilizzare l’ibridazione di un oligonucleotide di 25 basi alla sua sequenza bersaglio, nelle condizioni di temperatura e forza ionica raccomandate per l’ibridazione, solo il set di sonde PM sarà capace d’ibridare con la sequenza bersaglio, mentre le sonde MM non potranno farlo. Questa strategia nella progettazione delle sonde permetterà, al momento della lettura del microarray, di discriminare molto efficacemente tra segnale e rumore di fondo. Come conseguenza di questo complesso disegno, questo tipo di microarray offre contemporaneamente una grande specificità, dovuta alla molteplicità di sonde per ciascun trascritto, insieme a un’elevata sensibilità, dovuta a un’efficiente sottrazione del rumore di fondo. Inoltre, l’uso di sonde multiple per ciascun trascritto permette anche un’analisi statistica dei dati sperimentali, fornendo anche valori di confidenza e di probabilità.
Una tecnologia diversa, ma altrettanto efficace, è rappresentata da microarray le cui feature sono costituite da sonde della lunghezza di 60 nucleotidi, depositati sul chip utilizzando una tecnica di sintesi chimica in situ derivata dalla stampa a getto di inchiostro, senza contatto fisico col supporto. Questa tecnologia (introdotta dalla Agilent Technologies di Palo Alto) permette una grande flessibilità nella progettazione e realizzazione dei microarray, perché la loro costruzione è completamente controllata da software e non richiede la delicata preparazione di maschere fotolitografiche. Quindi è possibile ottenere serie limitate di microarray progettati e costruiti su misura. Inoltre l’uso di sonde lunghe 60 basi aumenta la specificità e la sensibilità, e permette una progettazione più semplice. Il software usato per la progettazione, infatti, riesce più rapidamente a selezionare sonde con una composizione di basi ottimale e contemporaneamente, confrontandole con le basi di dati genomiche, con un limitato potenziale di cross-ibridazione.
Esistono altre tre tipologie di microarray tecnologicamente e commercialmente importanti: la tecnologia BeadArray della ditta Illumina, basata su microsfere di 3 micron di diametro, ricoperte di oligonucleotidi che funzionano come sonde e marcatori, che si autoassemblano in maniera casuale in pozzetti ricavati nel substrato del microarray; la tecnologia CodeLink (Applied Microarrays) per costruire i Gene Expression Bioarray, microarray di oligonucleotidi legati al supporto tramite una matrice di gel tridimensionale; e la piattaforma della Applied Biosystems) che costruisce microarray di oligonucleotidi e visualizza le sequenze bersaglio ibridate con una tecnologia basata su chemiluminescenza.
Nel febbraio 2005, la FDA (Food and Drug Administration) statunitense ha lanciato il progetto MAQC (MicroArray Quality Control project), un’iniziativa della comunità scientifica promossa per rispondere alle preoccupazioni sorte negli ultimi anni concernenti l’affidabilità di questa tecnologia, sempre più diffusa e importante per le misure di espressione genica nella ricerca, nell’industria e anche in clinica. La FDA ha riconosciuto che i microarray rappresentano una tecnologia chiave in farmacogenomica e in tossicogenomica; tuttavia, prima che questa tecnologia possa essere usata con successo e sicurezza nella pratica clinica e nella creazione di norme regolamentari, ha ritenuto necessario sviluppare procedure standard e misure di qualità.
Il progetto MAQC è nato per fornire questi strumenti e per determinare obiettivamente: le capacità delle diverse piattaforme tecnologiche; le competenze dei diversi laboratori e i pregi e i difetti delle diverse procedure di analisi dei dati. Per realizzarlo, quattro diverse miscele di due distinti RNA di riferimento sono state analizzate in differenti laboratori usando sei piattaforme commerciali diverse di microarray e tre piattaforme tecnologiche alternative (basate principalmente su PCR quantitativa).
Nel settembre 2006, la rivista “Nature Biotechnology” ha pubblicato i risultati della prima parte del progetto. I dati, piuttosto incoraggianti nel complesso, hanno dimostrato (a) che la tecnologia dei microarray possiede le capacità richieste perché possa essere utilizzata per compiti normativi; (b) che i risultati sono ripetibili all’interno dello stesso laboratorio, riproducibili tra diversi laboratori e comparabili tra le diverse piattaforme tecnologiche; (c) che bisogna migliorare le annotazioni e la mappatura delle sonde per rendere più accurati i paragoni fra diverse piattaforme; (d) che i metodi di analisi influiscono sulla riproducibilità delle liste dei geni espressi differenzialmente; (e) che le piattaforme differiscono in robustezza e i laboratori in affidabilità.
Metodologia sperimentale
L’approccio tecnologico dei microarray per l’analisi dei profili di espressione genica si basa sulla proprietà dell’RNA o del DNA di ibridare alle proprie sequenze complementari. Come regola generale, le sequenze bersaglio marcate, costituite da DNA complementare (cDNA) preparato con l’enzima trascrittasi inversa dalla popolazione di RNA messaggero (mRNA) che si vuole esaminare, sono ibridate a sonde geniche specifiche distribuite e fissate su un supporto solido, il microarray. In queste condizioni, la quantità di sequenze bersaglio ibridate a ciascuna specifica sonda genica immobilizzata è proporzionale alla quantità di trascritto del gene corrispondente nella popolazione di mRNA originale.
Da un punto di vista operativo, i microarray ad alta densità si distinguono in due tipi: piattaforme di ibridazione con fluorescenza a due canali o a singolo canale.
Nel caso delle piattaforme di ibridazione che utilizzano la fluorescenza a due canali, a ciascun microarray sono ibridate simultaneamente uguali quantità di sequenze bersaglio provenienti da due campioni differenti, marcate con due fluorocromi differenti, in genere la cianina 3 (Cy3) e la cianina 5 (Cy5). I due campioni ibrideranno competitivamente con le sonde fissate al supporto del microarray e il rapporto delle quantità dei due fluorocromi ibridati a ciascuna feature sarà considerato uguale al rapporto delle quantità di quel trascritto specifico nei due campioni. L’uso del rapporto di ibridazione tra due campioni semplifica efficacemente molti problemi che derivano da disomogeneità nella fabbricazione e nell’ibridazione dei microarray e da differenze nella trascrizione inversa e marcatura delle sequenze bersaglio. Questa tecnologia è utilizzata soprattutto con i microarray basati su sequenze di cDNA stampate sul microarray per contatto.
Nel caso dell’ibridazione con fluorescenza a singolo canale, un unico campione è ibridato a ciascun microarray. I valori che si ottengono sono quindi valori assoluti, relativi a un singolo campione per volta. L’accuratezza, la specificità e la riproducibilità dei dati così ottenuti si basano sui seguenti fattori: l’alta precisione della tecnica usata nella produzione dei microarray; le condizioni di ibridazione estremamente controllate; le strategie stringenti per la selezione delle sonde e la logica altamente sofisticata dei controlli interni integrati nella progettazione dei microarray. Attualmente, soltanto i fabbricanti tecnologicamente più avanzati commercializzano questo tipo di piattaforma; prima fra tutti Affymetrix, che con i suoi GeneChip® è stata tra i pionieri di questa tecnologia, una delle più diffuse, soprattutto nel settore industriale.
Il principale vantaggio di questa tecnologia consiste nella possibilità di ottenere non soltanto dati comparativi, ma anche delle misurazioni assolute di espressione genica, eliminando la necessità di introdurre campioni di controllo in ogni esperimento, così come richiesto dalle piattaforme a due canali. La quantificazione assoluta dell’espressione genica permette di paragonare più facilmente campioni diversi, esaminati in luoghi e in tempi diversi, e apre la strada all’implementazione di grandi basi di dati che possano essere integrati con dati prodotti in differenti laboratori e interrogati indipendentemente da differenti ricercatori.
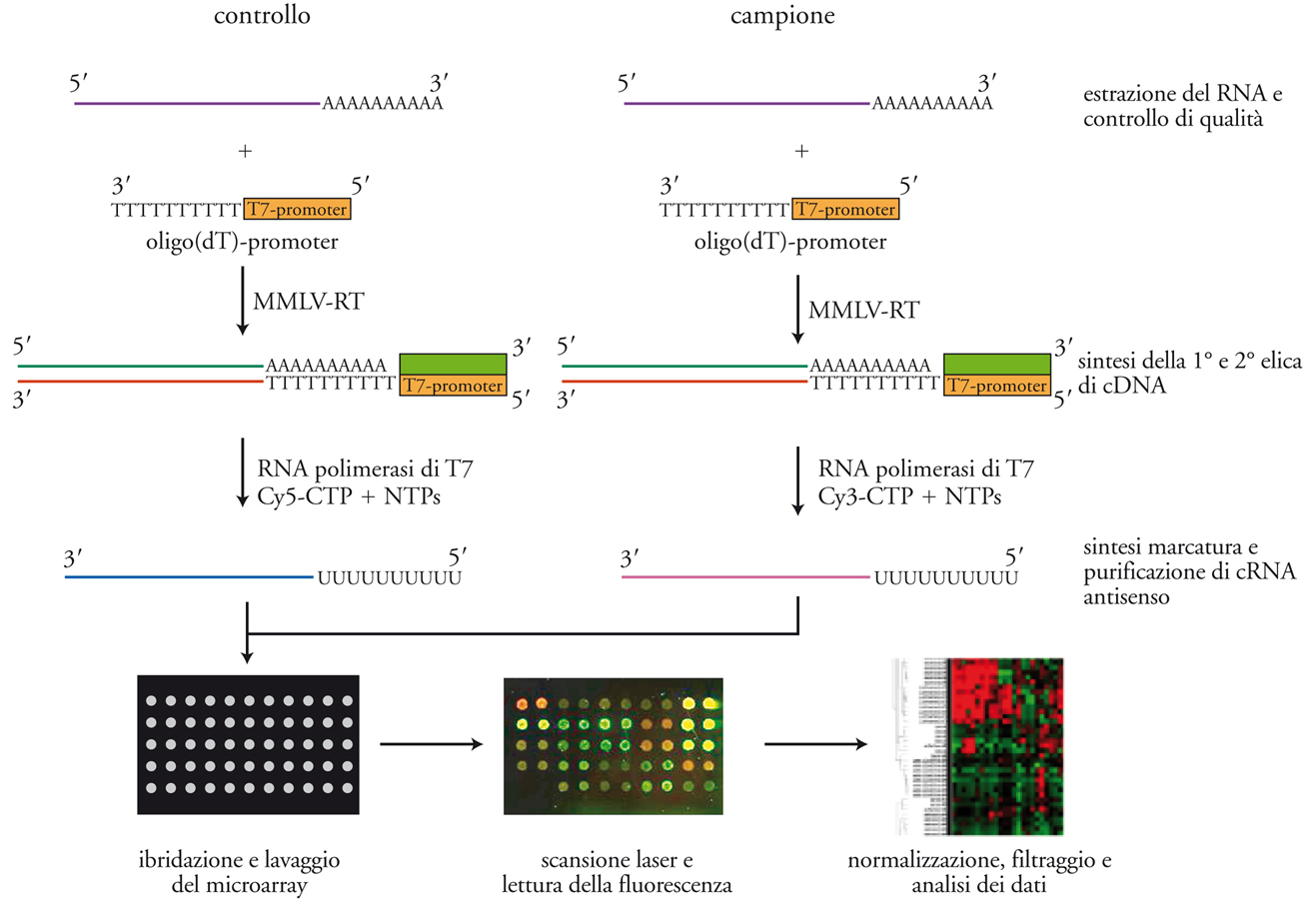
Un esperimento di analisi di profili di espressione genica tramite microarray si può dividere tipicamente in quattro fasi: (a) preparazione dei campioni e marcatura; (b) ibridazione e lavaggio; (c) acquisizione dei dati; (d) analisi dei dati. Nella fig. 3 è schematizzato il diagramma di flusso di un esperimento di questo tipo.
Preparazione dei campioni e marcatura
La preparazione dei campioni inizia con l’isolamento di RNA totale dalle cellule o dai tessuti in esame utilizzando metodi standard di biologia molecolare. L’estrazione deve essere completa e rappresentativa, in quanto la popolazione di mRNA presenti in questo RNA idealmente rappresenta, qualitativamente e quantitativamente, tutti i geni espressi nel campione biologico al momento dell’acquisizione. Particolarmente importante è il controllo del grado di purezza e di integrità della preparazione, perché il successo dell’esperimento dipende in gran parte dalla qualità dell’RNA. Solo le preparazioni di RNA che superano il controllo qualità potranno essere analizzate. Una contaminazione del campione da parte di proteine, lipidi e carboidrati aumenterà significativamente il rumore di fondo, perché questi composti possono mediare legami non specifici del cDNA marcato con le superfici di supporto del microarray. Di contro, una degradazione dell’RNA potrà essere causa di una non rappresentatività del campione.
Per ognuno dei campioni da esaminare, il messaggero contenuto nella preparazione di RNA totale sarà convertito in DNA complementare (cDNA) utilizzando l’enzima trascrittasi inversa e una corta sequenza innesco (primer sequence) costituita da oligo(dT), una breve sequenza di deossi-timina. Questa sequenza si accoppierà con la coda poliadenilata degli RNA messaggeri e funzionerà da innesco per la trascrittasi inversa che così convertirà in cDNA soltanto i messaggeri poliadenilati. Il cDNA così ottenuto può essere usato direttamente come campione per l’ibridazione, o può essere sottoposto a un ulteriore passaggio ed essere trascritto in RNA complementare(cRNA) da una RNA polimerasi. Quest’ultimo passaggio permette un’ulteriore amplificazione del campione e inoltre migliora l’efficienza di ibridazione in quanto gli ibridi DNA-RNA sono più stabili degli ibridi DNA-DNA. La marcatura fluorescente dei campioni di cDNA ocRNA in genere avviene per incorporazione enzimatica da parte della trascrittasi inversa o della polimerasi di un nucleotide precursore marcato con un fluorocromo. Nel caso di piattaforme a due canali, il campione e il relativo controllo sono marcati con due diverse molecole traccianti (in genere le cianine Cy3 e Cy5); nel caso di piattaforme a singolo canale tutti i campioni sono marcati con lo stesso fluorocromo.
Ibridazione e lavaggio
L’ibridazione è il processo tramite cui due filamenti complementari di acidi nucleici si accoppiano per formare una molecola a doppio filamento. I campioni marcati sono disciolti in una apposita soluzione per ibridazione, denaturati a temperatura elevata e quindi posti a contatto con il microarray in una camera da ibridazione di piccolo volume. Il volume ridotto della soluzione di ibridazione contribuisce a mantenere alta la concentrazione delle sequenze bersaglio e quindi ad aumentare la velocità di ibridazione. L’andamento cinetico della reazione di ibridazione è dipendente non solo dalla concentrazione delle sequenze bersaglio ma anche dalla forza ionica della soluzione, dalla temperatura e dalla durata della reazione. Tutti questi parametri sono cruciali per la riproducibilità dell’esperimento e, una volta determinati, devono essere mantenuti assolutamente costanti in esperimenti diversi.
Molti protocolli sperimentali prevedono anche un pretrattamento dei microarray prima dell’ibridazione, per denaturare le sonde e per saturare le superfici con molecole neutre e ridurre il rumore di fondo dovuto a legami non specifici.
I microarray sono lavati dopo l’ibridazione sia per rimuovere l’eccesso di sequenze bersaglio marcate che non hanno ibridato con le sonde, sia per aumentare la specificità dell’esperimento e ridurre l’ibridazione non specifica. Questo effetto si ottiene riducendo la forza ionica e aumentando la temperatura della soluzione di lavaggio.
Acquisizione dei dati
Alla fine del lavaggio, i microarray sono asciugati e si passa alla fase di acquisizione dei dati. A tale scopo bisogna ottenere un’immagine accurata della superficie ibridata del microarray, utilizzando uno scanner laser, a uno o a due colori, secondo la tecnologia usata. Il fascio del laser scansiona la superficie del microarray, eccitando i fluorocromi incorporati nelle sequenze bersaglio ibridate alle differenti feature e l’emissione fluorescente prodotta, di lunghezza d’onda caratteristica, viene raccolta e misurata da un’ottica da microscopio confocale. I dati raccolti sono trasformati in un’immagine digitale per ciascuna delle lunghezze d’onda eccitatrici. Queste immagini sono quindi esaminate con software appositi che estraggono i dati in un formato numerico utilizzabile per le successive analisi. Questi programmi svolgono diverse funzioni: (a) analizzano l’immagine e localizzano le feature, allineandole con la griglia teorica che descrive la posizione delle feature sulla superficie del microarray; (b) utilizzando le informazioni fornite dal fabbricante del microarray, assegnano alla singola feature tutte le annotazioni relative alla sonda in essa contenuta (identificazione e sequenza della sonda, simbolo del relativo gene, numero di accesso GenBank, ecc.); (c) misurano la quantità di sequenze bersaglio legate alla singola feature; (d) misurano il rumore di fondo locale nelle vicinanze di ciascuna feature; (e) combinano, nel caso di piattaforme a due canali, i dati ottenuti da scansioni del microarray con laser di diversa lunghezza d’onda; (f) esportano i dati ottenuti sia sotto forma di tabella numerica che sotto forma di immagine in falsi colori del microarray, perché possano essere ulteriormente analizzati.
A queste funzioni di base molti programmi associano una completa analisi statistica a livello della singola feature, esaminando in alcuni casi più di 100 parametridiversi. Per esempio, per ciascuna feature si tiene conto di varie distribuzioni statistiche dei pixel che la costituiscono, della forma e dell’uniformità del segnale, dellapresenza di pixel saturati, del rapporto segnale/rumore, e così via. In tal modo si ottengono informazioni chepossono essere usate per scartare o correggere i valorid’ibridazione di singole feature o per correggere globalmente i dati con vari tipi di normalizzazione.
Un’altra caratteristica che i programmi di acquisizione dei dati dovrebbero possedere, è la possibilità di esportare i dati nel formato MAGE-ML (MicroArray Gene Expression - Markup Language), compatibile con le basi di dati pubbliche che si propongono di conservare dati di trascrittomica in accordo con le raccomandazioni della Microarray and Gene Expression Data Society (http://www.mged.org/).
Analisi dei dati
Questa è la fase più complessa e meno standard della tecnologia dei microarray. Una volta estratti, in genere i dati sono filtrati per eliminare gli effetti di difetti del microarray o dell’ibridazione. Quindi, per paragonare i microarray fra loro, si ricorre a uno dei vari tipi di normalizzazione possibili, a seconda che si sia usata una piattaforma ad uno o a due canali. A questo punto, possibilmente basandosi su un’analisi di significatività statistica di repliche di dati, si ottiene la lista dei geni espressi differenzialmente nelle varie condizioni sperimentali studiate e di quelli la cui espressione è aumentata o diminuita. Importante adesso è la validazione dei dati ottenuti, cioè la conferma sperimentale di almeno parte dei dati con metodi diversi, quali la RT-PCR quantitativa oppure il Northern blot.
Si passa poi all’analisi esplorativa dei dati, con l’intento di trovare nei dati nuove configurazioni non derivabili dalle nozioni o dai preconcetti dello sperimentatore (unsupervised data analysis). Alcuni obiettivi tipici di quest’analisi sono l’identificazione di gruppi di geni la cui espressione abbia un andamento strettamente correlato nei vari campioni, o trovare sottogruppi ignoti nella popolazione dei campioni. Per rispondere a queste domande si usano metodi quali la cluster analysis o la principal components analysis.
Un altro tipo di approccio che sta guadagnando spazio è l’analisi orientata ai networks e ai pathways. I geni non agiscono mai da soli, ma sono integrati in una cascata di network di regolazione e di funzione. Analizzare i dati ottenuti dai microarray in una prospettiva di processi biologici interconnessi si sta affermando come il vero valore aggiunto di questo approccio tecnologico. Fondamentali per questo tipo di analisi si stanno rivelando i progetti di classificazione delle funzioni dei geni e delle loro interrelazioni quali per esempio il Gene ontology project (GO, http://www.geneontology.org/). Il progetto GO classifica i prodotti dei geni secondo tre concetti chiave: la loro funzione molecolare, il loro ruolo nei processi biologici multilivello, e la loro localizzazione nei componenti cellulari. Queste tre ontologie sono continuamente aggiornate e rese disponibili per essere utilizzate dai software di analisi.
Esistono molti software prodotti per visualizzare i dati dei microarray e analizzarli. Questi strumenti sono usati per scavare nei dati e rendere evidenti i cambiamenti di espressione genica più significativi statisticamente. Inoltre, la visualizzazione grafica facilita e rende più intuitiva l’analisi dei dati. Questi software hanno molte caratteristiche operative utili, come l’integrazione dei dati, la possibilità di filtrarli secondo diversi parametri, la capacità di riconoscere particolari configurazioni nei dati, di applicare agli stessi dei modelli interpretativi. Convenzionalmente, questi software usano il colore verde per indicare i geni la cui espressione è ridotta rispetto al controllo e il colore rosso per caratterizzare invece quelli la cui espressione è aumentata. In questo modo possono produrre delle mappe colorate che aiutano a visualizzare i dati di espressione.
Applicazioni della tecnologia
I microarray sono usati principalmente in due ambiti tecnologici: (a) per determinare il profilo di espressione genica di un tessuto o un organismo, cioè misurare la quantità dei trascritti dei diversi geni espressi in un certo momento in un campione biologico; (b) per identificare la presenza di specifiche sequenze geniche e di alterazioni e mutazioni di geni in un campione biologico.
Profili di espressione genica
Attualmente è probabilmente l’applicazione dei microarray più usata nell’ambito della ricerca biologica. Il profilo di espressione genica descrive qualitativamente e quantitativamente l’insieme dei geni trascritti in un dato momento da una cellula o da un tessuto. L’assunzione implicita è che il livello di trascrizione di ciascun gene rappresenti la risposta cellulare a uno stato particolare. I microarray disponibili ora possono fornire profili di espressione genica che riflettono la risposta trascrizionale di migliaia di geni a uno stimolo farmacologico o a un cambiamento dello stato cellulare. Tipicamente lo scopo che ci si propone è identificare nuovi geni coinvolti in un processo biologico oppure nuovi marcatori diagnostici/prognostici caratteristici di uno stato patologico.
L’approccio sperimentale dei microarray permette una ricognizione a livello dell’intero genoma con un singolo saggio, senza la necessità di formulare un’ipotesi a priori sui geni coinvolti nel fenomeno studiato. I profili di espressione genica prodotti dall’esperimento con i microarray potranno poi servire da base per identificare geni candidati per un successivo studio utilizzando le tecniche d’indagine tradizionali della biologia molecolare (Northern blot, Western blot, RT-PCR, transfezione genica ecc.). L’utilità di quest’approccio sperimentale è stata dimostrata già da numerose pubblicazioni e di seguito sono elencate le principali utilizzazioni della tecnologia.
Il profilo di espressione per predire la funzione di geni. - I profili di espressione possono rivelare correlazioni nell’espressione dei geni che possono essere utilizzate per predire la funzione del prodotto di un gene. I geni possono essere raggruppati in blocchi (clusters) con profili di espressione simili in varie condizioni sperimentali. Quest’operazione può essere fatta manualmente o con software appositi usando metodi statistici. Ci si aspetta che i geni che mostrano un andamento simile in diverse condizioni possano essere correlati funzionalmente. In uno studio precursore e fondamentale per la diffusione della tecnica dei microarray, il gruppo di ricerca di Joseph DeRisi ha esaminato a livello dell’intero genoma l’espressione genica del microrganismo Saccharomyces cerevisiae durante il passaggio dal metabolismo fermentativo anaerobico a quello respiratorio aerobico. Per mezzo di algoritmi di clustering, gli autori hanno identificato schemi temporali di induzione e repressione ed hanno raggruppato i geni secondo la similarità dei loro profili di espressione. In molti casi questi raggruppamenti coincidevano con raggruppamenti funzionali. Per esempio, i geni correlati al citocromo C e quelli del ciclo TCA/glioxilato e dell’immagazzinamento dei carboidrati erano indotti dall’esaurimento del glucosio mentre quelli relativi alla sintesi delle proteine ribosomiali erano repressi.
In altri casi, i gruppi mostravano meccanismi di regolazione trascrizionale identici. Per esempio sette geni condividevano un’induzione precoce con un picco di espressione tardiva a 18,5 ore. Si scoprì che tutti questi sette geni possedevano a monte del punto di inizio della trascrizione una sequenza di regolazione chiamata elemento di risposta allo stress (STRE). Esaminando con attenzione altri tredici geni che mostravano lo stesso profilo di espressione, fra cui dieci allora non ancora caratterizzati, si vide che ben nove di essi possedevano sequenze STRE.
Il profilo di espressione come ‘impronta digitale’ o ‘firma’. - Il profilo di espressione ottenuto da un microarray può essere considerato la fotografia della risposta trascrizionale di una cellula a una situazione fisiologica o patologica. Questa risposta è in genere regolata in modo rigoroso e riproducibile in funzione dello stimolo che l’ha originata. Il profilo di espressione può quindi essere considerato una specie d’impronta digitale di una cellula o tessuto in una determinata situazione. Questa impronta molecolare potrà essere utilizzata per riconoscere lo stato metabolico di una cellula o per classificare un tipo di patologia. Per esempio, i tumori di uno stesso tipo istologico possono essere classificati anche in base ai loro profili di espressione genica. Questi profili a loro volta possono essere correlati a dati concernenti la sopravvivenza dei pazienti, la capacità del tumore primario di creare metastasi, la risposta a determinati farmaci ecc. Questo tipo di studi ha portato allo sviluppo di signatures (firme), gruppi ridotti di geni il cui profilo di espressione considerato complessivamente caratterizza o uno stato patologico preciso, o la capacità di rispondere a un farmaco, oppure la probabilità di andare incontro a recidive.
Recentemente, sono state pubblicate due signature particolarmente importanti, per la prognosi del tumore del seno linfonodo-negativo (LNN), chiamate Amsterdam 70-gene e Rotterdam 76-gene signatures, dal numero dei geni che considerano e dalla città in cui sono state sviluppate. Entrambi gli strumenti sono studiati per definire fra donne colpite da tumore LNN, quali hanno la più alta probabilità di essere curate totalmente dalla sola chirurgia e quali invece hanno un alto rischio di recidiva e di sviluppo di metastasi distanti nel corso dei successivi cinque anni. I due test hanno dimostrato di avere un’accuratezza di più del 90%. Adesso circa l’80% di queste pazienti viene trattato con chemioterapia, mentre il 60÷70% potrebbe essere curato efficacemente solo dalla chirurgia. L’uso di questi test potrebbe quindi avere un impatto molto importante sulle terapie da somministrare e sulla qualità di vita delle pazienti.
I profili di espressione genica nello sviluppo di farmaci. - Dato che il processo di scoperta, sperimentazione e validazione di nuovi farmaci normalmente dura molti anni e ha un costo molto elevato, e che solo pochi dei farmaci candidati superano lo stadio della validazione, qualsiasi metodo capace di migliorare l’efficienza del processo e di aumentare le probabilità di sviluppare farmaci efficaci è ben accetto da parte dell’industria farmaceutica. I microarray sono in grado di fornire informazioni utili a tutti gli stadi del processo di sviluppo di un farmaco. L’identificazione dei potenziali bersagli dell’azione dei farmaci può essere facilitata dalla migliore comprensione dei processi metabolici che si può ottenere dallo studio dei geni co-espressi. La proteina bersaglio di un farmaco può essere identificata trovando il gene che causa gli stessi cambiamenti del farmaco qualora venga rimosso dalla cellula o dall’organismo. In seguito, una volta che i farmaci candidati siano stati identificati e selezionati, i microarray possono essere usati per definire le loro proprietà tossiche esaminando i profili di espressione genica indotti dal trattamento con i farmaci. D’altra parte, molte funzioni di farmaci sono state identificate sulla base dei cambiamenti dell’espressione genica che essi provocano.
Identificazione di specifiche sequenze geniche
L’identificazione di specifiche sequenze geniche, in forma normale o alterata, in un campione biologico, è il secondo tipo di applicazione per cui è usata la tecnologia dell’ibridazione su microarray. Di seguito sono elencate le principali utilizzazioni.
Ibridazione genomica comparativa basata su microarray (aCGH, Array comparative genomic hybridization). - È noto che riarrangiamenti cromosomici sono comunemente associati a molte situazioni patologiche, tra cui vari tipi di cancro e molte malattie genetiche dello sviluppo, tra cui la sindrome di Down e la sindrome di Mowat-Wilson. L’identificazione e l’analisi di questi riarrangiamenti sono stati essenziali per lo studio di tali malattie e attualmente rappresentano preziosi e diffusi metodi di diagnosi clinica. Finora, la tecnica più usata per identificare queste variazioni cromosomiche è stata l’ibridazione genomica comparativa convenzionale (CGH), tecnologia derivata dalla FISH (Fluorescence in-situ hybridization). Il principio della tecnica si basa sull’ibridazione competitiva a cromosomi in metafase provenienti da un soggetto sano del DNA del paziente in esame e di un DNA di riferimento, anch’esso proveniente da un soggetto sano. I due DNA sono marcati con due diversi fluorocromi, rosso e verde, rispettivamente. Dopo l’ibridazione, un software apposito riconosce i singoli cromosomi e misura le intensità relative delle fluorescenze rosso e verde sull’intera lunghezza dei diversi cromosomi, calcolandone il rapporto. Una duplicazione o una delezione di un’area del genoma in esame si riflette in una variazione del rapporto delle fluorescenze in una specifica zona di un cromosoma. È così possibile riconoscere il tipo di difetto, delezione o amplificazione genica, e localizzare la regione cromosomica coinvolta. Questa tecnica tuttavia è molto laboriosa e necessita di personale altamente addestrato e specializzato in citogenetica. Un altro limite della CGH tradizionale è la bassa risoluzione dell’analisi genomica che fornisce, rendendo possibile solo l’identificazione di grosse aberrazioni cromosomiche, più grandi di 2 Mb (1 Mb = 1 megabase = 1 milione di basi di DNA) per quanto riguarda le amplificazioni e di 5-10 Mb per le delezioni.
L’introduzione della CGH basata su microarray (aCGH) ha rappresentato un grande progresso per questa tecnologia. L’aCGH permette di identificare alterazioni cromosomali (delezioni, amplificazioni e microamplificazioni) a livello dell’intero genoma. I microarray CGH sono costituiti da feature che contengono sonde localizzate lungo tutto il genoma. La risoluzione del microarray dipende dalla spaziatura delle sonde. Se un array ha una risoluzione di 1 MB, significa che ha approssimativamente una sonda per megabase. Le sonde possono essere preparate tramite PCR da cloni di cDNA o da cloni BAC. Più recentemente sono stati introdotti aCGH array con sonde formate da lunghi oligonucleotidi sintetici. In questo caso, gli array CGH possono essere costruiti usando un insieme di frammenti di DNA che si affiancano o addirittura si sovrappongono per coprire tutto il genoma o solo parte di esso, ad altissima risoluzione.
Tecnicamente la procedura d’ibridazione è simile a quella della CGH classica. Il DNA genomico da esaminare e un DNA genomico di riferimento sono marcati, rispettivamente con un fluorocromo verde e uno rosso. Entrambi i DNA sono ibridati contemporaneamente alle feature poste sul microarray, che coprono tutto il genoma o solo una parte di esso, a una maggiore risoluzione. Al termine dell’ibridazione e dopo i lavaggi per eliminare l’eccesso di DNA che non ha reagito, i microarray sono letti con uno scanner laser a due canali. Le immagini dei segnali fluorescenti sono catturate e analizzate, ed è calcolato il rapporto del segnale verde su quello rosso. In corrispondenza di ciascuna feature, un aumento del segnale rosso indicherà la presenza di una delezione nel DNA in esame, viceversa un aumento del segnale verde indicherà un’amplificazione. Il software poi trasformerà automaticamente questi risultati in una panoramica ad alta risoluzione delle delezioni e amplificazioni, affiancata alla rappresentazione grafica dei cromosomi, con l’indicazione dei loci e dei geni coinvolti e molte altre annotazioni utili.
ChIP on Chip. Dietro questo gioco di parole si nasconde una tecnica molto potente, che ha sempre maggiori utilizzazioni nella ricerca della biologia moderna. Il nome per esteso significa Chromatin immuno precipitation on microarray Chip, cioè immunoprecipitazione della cromatina analizzata su microarray. La cromatina è il complesso nucleoproteico formato dall’insieme del DNA genomico e le proteine che lo legano e lo organizzano, regolandone così la funzione replicativa e trascrizionale. Fra queste proteine ci sono: istoni, fattori e cofattori trascrizionali, enzimi di modificazione, di trascrizione e di replicazione del DNA, e così via.
La tecnica ChIP on Chip permette di determinare la presenza e la posizione in una specifica parte del genoma di un particolare tipo di proteina, per esempio di un fattore trascrizionale. Semplificando, la domanda cui si può rispondere utilizzando questa tecnica è quali siano i geni il cui promotore è regolato da uno specifico fattore trascrizionale in determinate condizioni fisiologiche o patologiche. La procedura consiste nel fissare e isolare la cromatina delle cellule che si vogliono esaminare. La fissazione blocca, in maniera reversibile, le proteine legate al DNA al momento dell’esperimento. La cromatina è quindi frammentata in modo regolare con ultrasuoni e sottoposta a immunoprecipitazione utilizzando un anticorpo che riconosce il fattore trascrizionale che si desidera localizzare. L’immunoprecipitazione recupererà solo i frammenti di cromatina in cui è presente il fattore trascrizionale in esame. Il DNA presente in questi frammenti quindi conterrà le sequenze genomiche riconosciute nelle condizioni sperimentali prescelte da questo fattore trascrizionale. Questo DNA è estratto, marcato con un fluorocromo e quindi ibridato a un microarray dello stesso tipo di quelli utilizzati per la tecnica aCGH. In questo caso, dopo l’ibridazione mostreranno un segnale fluorescente solo le feature corrispondenti alle regioni del genoma a cui era legato il fattore trascrizionale. Questa tecnica permette quindi di identificare con alta risoluzione le regioni genomiche importanti per i diversi processi di regolazione trascrizionale.
Identificazione dei polimorfismi a singolo nucleotide (SNP) e diagnosi. - I polimorfismi a singolo nucleotide (Single nucleotide polymorphism, SNP), variazioni in un singolo sito nel DNA, rappresentano le alterazioni genomiche più frequenti. Per esempio, nel genoma umano si stima la presenza di 5÷10 milioni di SNP. Poiché gli SNP sono molto conservati nel corso dell’evoluzione e all’interno della popolazione, essi possono essere utilizzati come perfetti marcatori genotipici. Attualmente, nelle banche dati pubbliche sono registrati più di 2 milioni di SNP con frequenze alleliche note. Individuare queste variazioni è molto utile perché è possibile associarle con la suscettibilità a malattie genetiche oppure con l’efficacia della risposta a farmaci dei singoli individui. Questo tipo di studi ora è in grande espansione e, presto, analizzando i polimorfismi della popolazione, i medici potranno compiere scelte terapeutiche su misura per il singolo paziente.
Già da qualche anno esistono tecniche basate sull’uso di microarray per analizzare gli SNP, e recentemente sono stati presentati metodi per estendere tale indagine al livello dell’intero genoma. Tali metodi, estremamente complessi, sono già disponibili commercialmente e quindi il loro impiego è in rapida espansione.
Basi di dati che raccolgono risultati ottenuticon microarray
L’enorme quantità di dati prodotta da un esperimento basato su microarray ha sollecitato la creazione di basi di dati di pubblico accesso destinati a raccoglierli. Infatti, molto spesso questi dati, che sono stati prodotti e analizzati in funzione di una specifica domanda scientifica, contengono ancora una grande quantità di informazioni non sfruttate. Questi stessi dati, raccolti in una base di dati e interrogati in modo adeguato, possono essere di grande utilità per altre ricerche.
Per facilitare l’archiviazione e lo scambio dei dati, nel 2001 è stato messo a punto uno standard per registrare gli esperimenti di espressione genica basati su microarray denominato MIAME (Minimum information about a microarray experiment). Le specifiche di questo standard prevedono la registrazione di tutte le informazioni necessarie a interpretare i risultati di un esperimento in modo non ambiguo e a riprodurre l’esperimento. Lo standard definisce il contenuto e la struttura delle informazioni considerate necessarie, piuttosto che il formato tecnico di archiviazione.
Sono stati creati tre archivi pubblici, in Europa, Stati Uniti e Giappone, con il compito di accettare, conservare, coordinare e distribuire i dati degli studi basati su microarray in formato compatibile con lo standard MIAME. Essi sono ArrayExpress presso l’European Bioinformatics Institute (Cambridge, UK; http:// www.ebi.ac.uk/arrayexpress/), GEO (Gene expression omnibus) presso il National Center for Biotechnology Information (NCBI, Bethesda, MD, USA; http://www.ncbi.nlm.nih.gov/geo/) e CIBEX (Center for Information Biology gene EXpression database) presso il DNA Data Bank of Japan (DDBJ, Mishima, Giappone; http://cibex.nig.ac.jp/index.jsp). Molte riviste scientifiche adesso esigono che, prima della pubblicazione, i dati che si riferiscono a esperimenti con microarray siano depositati in uno di questi archivi, in modo da renderli disponibili alla comunità scientifica.
Oltre a questi archivi principali, ne esistono altri creati per conservare dati relativi a specifici settori di studio. Per esempio, il caArray (Cancer Array Informatics Project) sviluppato dal Centro per la Bioinformatica del NCI (National Cancer Institute, USA), che consiste di una base di dati dedicata allo studio del cancro e di strumenti software progettati appositamente per essa (http://caarray.nci.nih.gov/). Oppure il PLEXdb (PLant EXpression DataBase), sviluppato dalla Iowa State University come risorsa pubblica specializzata nella conservazione e nell’interrogazione dei profili di espressione genica relativi alle piante e ai patogeni delle piante (http://www.plexdb.org/). O ancora, il GXD (Gene eXpression Database), presso il Jackson Laboratory (Bar Harbor, ME, USA), un archivio pubblico di informazioni di espressione genica dedicato al topo da laboratorio, che contiene in particolare dati relativi allo sviluppo, al differenziamento e all’anatomia (http://www.informatics.jax.org/mgihome/GXD/aboutGXD.shtml). Merita una menzione anche lo Stanford Microarray Database (SMD), che accoglie dati relativi a tutte le applicazioni dei microarray e dispone di strumenti software molto avanzati sviluppati appositamente dall’Università di Stanford per l’analisi dei dati (http://genome-www5.stanford.edu/).
Bibliografia
Chetverin, Kramer 1993: Chetverin, Alexander B. - Kramer, Fred R., Sequencing of pools of nucleic acids on oligonucleotide arrays, “Biosystems”, 30, 1993, pp. 215-231.
Cheung 1999: Cheung, Vivian G. e altri, Making and reading microarrays, “Nature genetics”, 21, 1999, pp. 15-19.
DeRisi 1997: DeRisi, Joseph L. - Iyer, Vishwanath R. - Brown, Patrick O., Exploring the metabolic and genetic controlof gene expression on a genomic scale, “Science”, 278, 1997, pp. 680-686.
Duggan 1999: Duggan, David J. e altri, Expression profiling using cDNA microarrays, “Nature genetics”, 21, 1999,pp. 10-14.
Lockhart 1996: Lockhart, David J. e altri, Expression monitoring by hybridization to high-density oligonucleotide arrays, “Nature biotechnology”, 14, 1996, pp. 1675-1680.
Pease 1994: Pease, Ann C. e altri, Light-generated oligonucleotide arrays for rapid DNA sequence analysis, “Proceedingsof the National Academy of Sciences USA”, 91, 1994,pp. 5022-5026.
Schena 1995: Schena, Mark e altri, Quantitative monitoring of gene expression patterns with a complementary DNA microarray, “Science”, 270, 1995, pp. 467-470.
Sellick 2004: Sellick, Gabrielle S. e altri, Genomewide link- age searches for Mendelian disease loci can be efficiently conducted using high-density SNP genotyping arrays, “Nucleic acids research”, 32, 2004, p. 164.
Syvänen 2005: Syvänen, Ann-Christine, Toward genome-wide SNP genotyping, “Nature genetics”, 37, 2005, S5-S10.