
Cellula. Matrice extracellulare
Cellula. Matrice extracellulare
La matrice extracellulare (MEC) o ECM (Extracellular matrix), come più comunemente viene definita nell'ambito scientifico internazionale, rappresenta la più complessa unità di organizzazione strutturale dei tessuti degli organismi viventi. I tessuti, infatti, non sono costituiti solo da cellule: una parte rilevante del loro volume è formata dallo spazio extracellulare, occupato da un'intricata rete di macromolecole, la cui organizzazione tridimensionale rappresenta appunto l'ECM. L'analisi biochimica dell'ECM rivela che essa è composta da una pletora di proteine e polisaccaridi, che si aggregano in un reticolo organizzato in maniera compatta e connesso alla superficie delle cellule che l'hanno prodotto e di quelle circostanti. Se fino a qualche tempo fa si pensava che l'ECM servisse principalmente da impalcatura relativamente inerte in grado di stabilizzare la struttura fisica dei tessuti, è ormai ampiamente dimostrato che l'ECM rappresenta il substrato su cui tutte le cellule dei tessuti possono aderire, migrare, proliferare e differenziare, e che ne influenza inoltre la sopravvivenza, la forma e la funzione. Infatti, le macromolecole dell'ECM sequestrano fattori di crescita, molecole come l'acqua o i minerali, e controllano fenomeni fisiologici, quali la morfogenesi, fisiopatologici, quali la guarigione delle ferite, e patologici, quali l'invasione e la metastatizzazione tumorale.

Sebbene l'ECM sia presente strutturalmente e funzionalmente in tutte le componenti tessutali che costituiscono un organo, quali epiteli, vasi, muscoli, nervi e connettivo, è proprio in quest'ultimo, dove è anche definita 'sostanza fondamentale', che essa è più abbondante delle cellule, tanto da determinare le proprietà dell'organo stesso. Se pensiamo che i tessuti connettivi, costituenti primari della cute e delle ossa, formano l'impalcatura degli organi, comprendiamo come sia proprio l'organizzazione quantitativa e qualitativa delle macromolecole della loro ECM a determinare la tipologia di tessuto connettivo più adatto ai requisiti funzionali dei vari organi: l'ECM può calcificare, come nei tessuti ossei, dove forma strutture solide come la roccia, o costituire la struttura trasparente della cornea, o assumere l'organizzazione che conferisce ai tendini la loro enorme resistenza alla trazione.
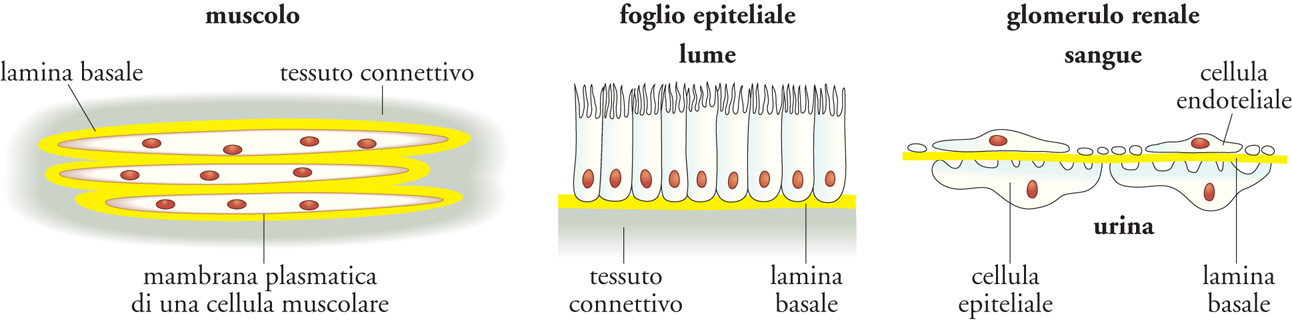
L'ECM non solo si presenta come sostanza extracellulare, ma è anche organizzata in strutture specializzate come le lamine basali (LB). Esse si trovano alla base di tutti gli epiteli e gli endoteli e circondano anche singole cellule muscolari, gli adipociti e le cellule di Schwann, che avvolgono gli assoni neuronali formando la mielina. Le LB giocano anche un ruolo importante nella rigenerazione dei tessuti dopo un danno. Quando tessuti come il muscolo, il nervo e l'epitelio vengono danneggiati, le LB sopravvivono e forniscono un'impalcatura lungo la quale le cellule in rigenerazione possono migrare.
Composizione molecolare dell'ECM
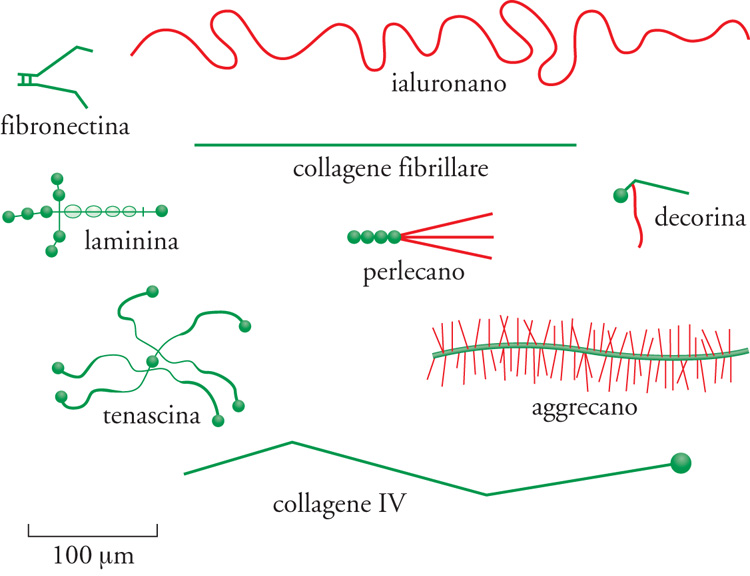
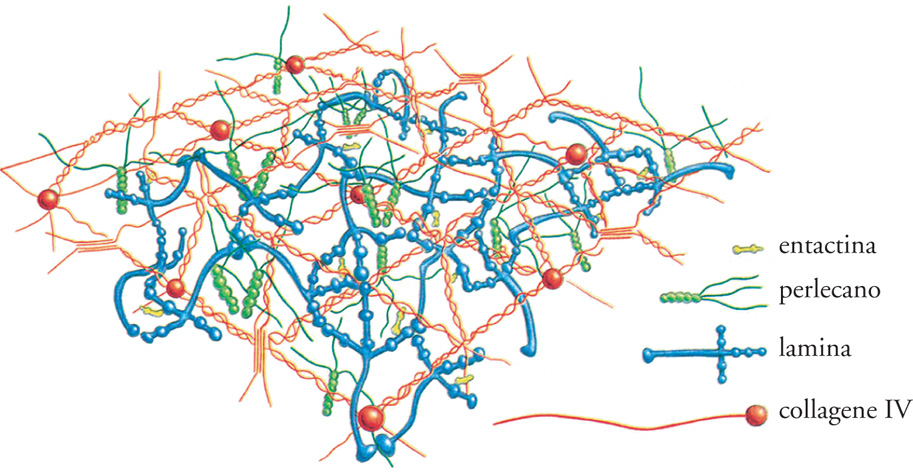
L'ECM (Extracellular matrix) è composta da due principali classi di macromolecole: catene polisaccaridiche appartenenti alla classe dei glicosamminoglicani (GAG) e proteine fibrose (fig. 2). Le prime si trovano solitamente legate alle proteine per formare i proteoglicani, che possono essere ricchi di gruppi solfato (il condroitinsolfato, il dermatansolfato, l'eparansolfato e il cheratansolfato) o privi (l'acido ialuronico). Le seconde comprendono due gruppi: uno con funzione principalmente strutturale (i collageni e l'elastina), e uno con funzioni principalmente adesive (la fibronectina, le laminine, le entactine o nidogeni e la vitronectina). Tutte queste macromolecole sono caratterizzate da grande varietà di forme e di dimensioni. Le molecole dei glicosamminoglicani e dei proteoglicani formano, nei tessuti connettivi, una sostanza 'fondamentale' gelatinosa e fortemente idratata, in cui sono immerse le proteine fibrose; questo gel di polisaccaridi consente la diffusione di sostanze nutritive, metaboliti e ormoni tra il sangue e le cellule dei tessuti e resiste alle forze compressive esercitate sull'ECM. Le fibre dei collageni rinforzano l'ECM, la organizzano e ne assicurano la resistenza alla trazione, mentre le fibre dell'elastina ne determinano l'elasticità. Alcuni collageni, interagendo con le laminine, le entactine o nidogeni e i proteoglicani perlecano e agrina, costituiscono le impalcature delle lamine basali, o LB (fig. 3 e fig. 4). Le proteine d'adesione agevolano inoltre la connessione delle cellule tessutali all'ECM stessa e ne influenzano la polarizzazione: la fibronectina, infatti, favorisce il congiungimento dei fibroblasti e di altre cellule con la matrice dei tessuti connettivi, mentre le laminine favoriscono quello delle cellule epiteliali con le LB. La vitronectina, infine, interagisce con l'elastina, i glicosamminoglicani e i collageni e modula l'angiogenesi e la degradazione dell'ECM stessa.
Oltre ai GAG e alle proteine fibrose strutturali e adesive sopra descritte, nell'ECM sono presenti anche altre proteine, definite 'proteine matricellulari'. Esse costituiscono una nuova classe di proteine di secrezione che non hanno funzioni strutturali, ma rivestono il ruolo di adattatori molecolari in quanto interagiscono con le proteine fibrose e adesive dell'ECM, con recettori cellulari o con altre molecole come fattori di crescita, citochine e proteasi. Sebbene diverse tra loro per funzione, le proteine matricellulari hanno in comune la capacità di modulare le interazioni cellula-matrice. Questa famiglia di proteine adattatrici comprende: (a) la SPARC (proteina secreta acida e ricca in cisteina), conosciuta anche come 'osteonectina', che contribuisce al rimodellamento dei tessuti in risposta al danno e funge da inibitore dell'angiogenesi; (b) le trombospondine, una famiglia di grandi proteine multifunzionali alcune delle quali, analogamente alla SPARC, inibiscono l'angiogenesi; (c) l'osteopontina, che regola la calcificazione ossea ma che agisce anche come promotore della migrazione dei leucociti; (d) i membri della famiglia della tenascina, grandi proteine multimeriche coinvolte nella morfogenesi e nella modulazione dell'adesione cellulare; (e) le matriline, principalmente espresse nella cartilagine, che partecipano alla formazione delle componenti fibrillari e filamentose dell'ECM; (f) le fibuline, che, associate a fibre elastiche, microfibrille di fibronectina, aggregati di proteoglicani e a componenti delle LB, sono coinvolte nello sviluppo embrionale e nell'organizzazione strutturale e funzionale dei tessuti cardiaci, cutanei e oculari; (g) le emiline che sono associate alle fibre elastiche come le fibuline e regolano l'elastogenesi e il mantenimento delle strutture vascolari. Le macromolecole che costituiscono l'ECM sono per lo più secrete localmente dalle cellule presenti nell'ECM: esse ne controllano anche l'organizzazione e l'orientamento. Nella maggior parte dei tessuti connettivi le macromolecole dell'ECM sono prodotte dai fibroblasti. Tuttavia, in alcuni tessuti connettivi specializzati, come la cartilagine e l'osso, esse sono secrete da fibroblasti specifici, quali i condroblasti e gli osteoblasti; infine, possono essere sintetizzate anche da cellule epiteliali, endoteliali e leucocitarie.
I principali recettori delle macromolecole dell'ECM: le integrine
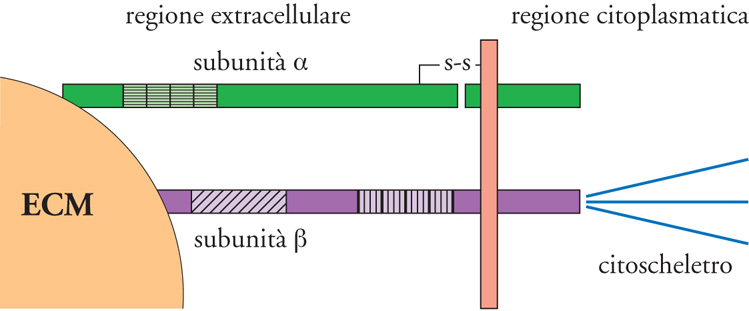
Gran parte delle macromolecole dell'ECM può stabilire interazioni altamente funzionali con le cellule di un tessuto grazie ai recettori che queste esprimono. Tra questi, i più importanti sono le integrine, in quanto rappresentano la via fondamentale con cui le cellule si legano all'ECM e rispondono ai suoi stimoli. Le integrine sono proteine eterodimeriche transmembranarie costituite da due subunità associate, indicate con le lettere greche α e β, che presentano entrambe un'estesa porzione extracellulare, un dominio transmembrana e una porzione intracitoplasmatica (fig. 5). Al momento sono state descritte più di 10 differenti catene β (β1, β2, β3, β4, ecc.) e più di 20 diverse subunità α (α1, α2, α3, α4, α5, α6, α7, ecc., con l'eccezione di due, definite αv e αE). Mentre le catene β hanno la possibilità di unirsi a numerose catene α, queste ultime si associano generalmente a una sola subunità β, a differenza della subunità αv che ha la capacità di legare diverse catene β come le β1, le β3, le α5, le α6, e le α8. Tutte queste complesse associazioni portano alla formazione di circa 30 differenti integrine, che sono quindi classificate in sottofamiglie aventi come denominatore comune la stessa catena β o la catena αv.
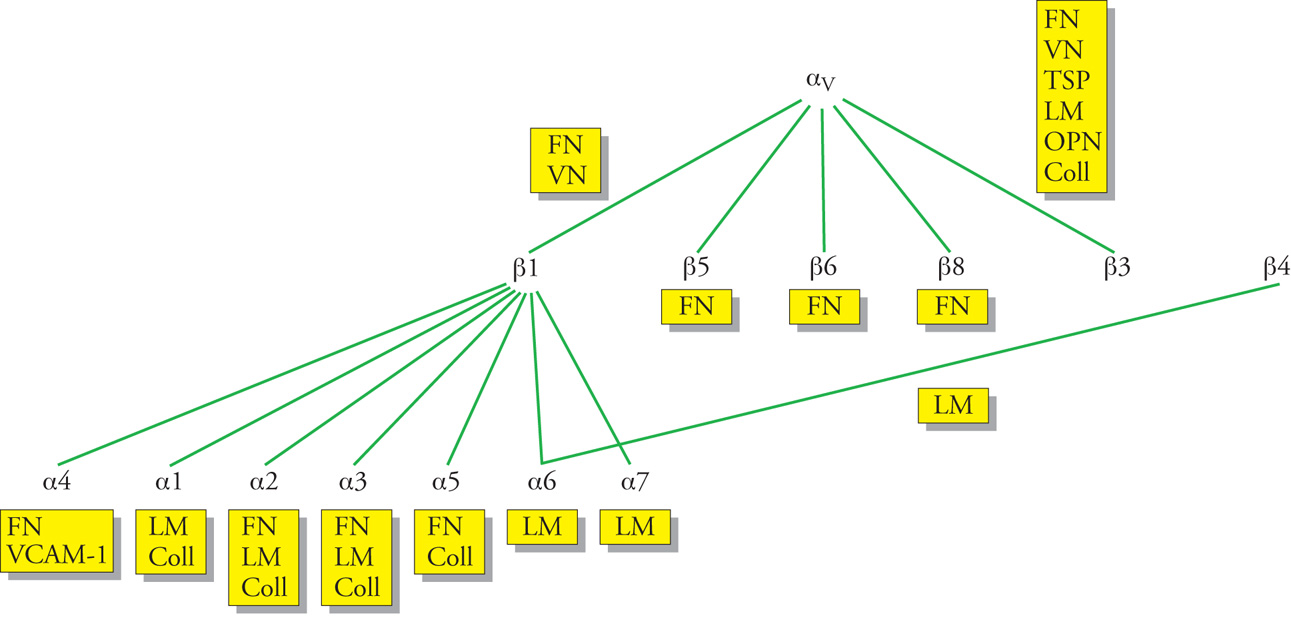
Le integrine β1, β3, β4 e αv sono 'classicamente' quelle in grado di riconoscere con le loro porzioni extracellulari le proteine dell'ECM. Nell'ambito della famiglia delle integrine β1, le integrine α1β1 e α2β1 sono entrambi recettori delle laminine e dei collageni, mentre l'α3β1 lega, oltre a questi, anche la fibronectina. L'α5β1 lega esclusivamente la fibronectina, mentre l'α6β1 e l'α7β1 sono i maggiori recettori delle laminine. A differenza di tutte queste integrine, l'α4β1, oltre a legare la fibronectina, è in grado di mediare anche interazioni cellula-cellula avendo la capacità di legare il VCAM-1 (molecola-1 di adesione cellulare vascolare), presente sulle cellule endoteliali attivate da citochine. Di tutte le integrine β3, l'αvβ3 è un recettore in grado di legare la vitronectina, la fibronectina, le trombospondine e l'osteopontina. L'integrina α6β4 è il maggiore recettore epiteliale delle laminine (fig. 6).
Alcune delle sequenze amminoacidiche delle regioni che le integrine riconoscono sulle proteine dell'ECM sono conosciute e caratterizzate: la sequenza GRGDSP, abbreviata come RGD, costituita dagli amminoacidi glicina - arginina - glicina - acido aspartico - serina - prolina, è presente su fibronectina e vitronectina e viene legata da α5β1, α3β1 e da altre integrine contenenti la subunità αv; la sequenza DGEA, formata dagli amminoacidi acido aspartico - glicina - acido glutammico - alanina, è presente nel collagene di tipo I ed è legata dall'eterodimero α2β1; la sequenza EILDV, composta dagli amminoacidi acido glutammico - isoleucina - leucina - acido aspartico- valina, è contenuta nella fibronectina ed è riconosciuta dal complesso α4β1.
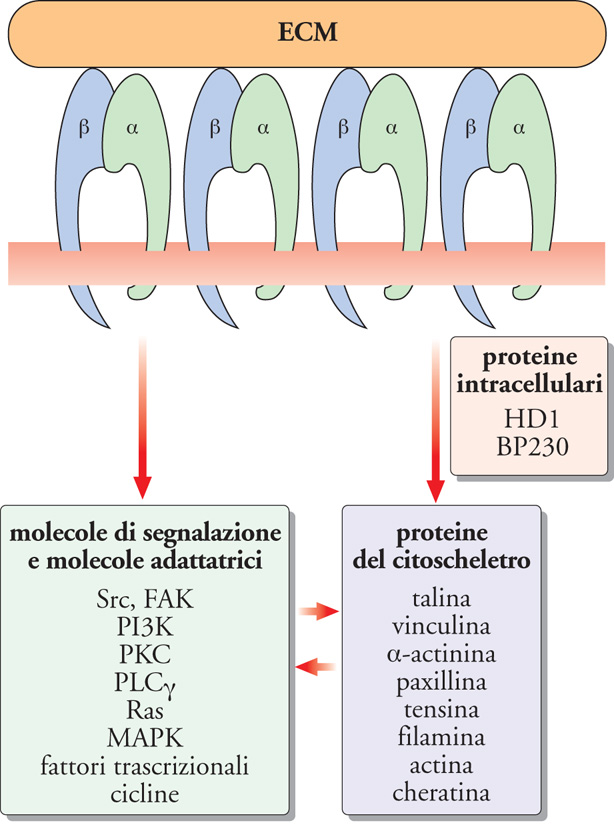
Le integrine assicurano anche le comunicazioni tra l'ECM e l'interno delle cellule, in quanto le porzioni intracitoplasmatiche delle loro subunità α e β interagiscono con proteine del citoscheletro e intracellulari. Le connessioni più rilevanti riguardano le subunità β. È stato dimostrato che la β1 lega la talina, la vinculina, la tensina e l'α-actinina, la β2 l'α-actinina, la paxillina e la filamina, la β3 l'α-actinina e la paxillina e la β4 proteine intracellulari, come l'HD1, la proteina 1 degli emidesmosomi (HD) nota anche come 'plectina', e il BP230, l'antigene del peso molecolare di 230 kDa del pemfigoide bolloso (BP). Tramite queste interazioni, le subunità β sono in connessione con lo scheletro di actina e di cheratina del citoplasma delle cellule (fig. 7).
Le integrine, quando legano i componenti dell'ECM, sono in grado inoltre di trasmettere segnali biochimici all'interno della cellula che le esprime. Recentemente è stato identificato un gran numero di mediatori dell'attivazione cellulare indotta dalle integrine: chinasi della famiglia Src (oncogene trasformante del virus del sarcoma di Rous) e della famiglia delle adesioni focali, il cui prototipo è FAK (chinasi delle adesioni focali); serino-treonino-chinasi, come la PKC (proteina-chinasi di tipo C) e le MAPK (proteine-chinasi attivate dai mitogeni); molecole adattatrici; proteine che legano il GTP (guanosina trisfosfato), tra cui quelle appartenenti alla famiglia Ras (oncogene trasformante associato al sarcoma di Ratto); la PLCγ (fosfolipasi Cγ) e la PI3-K (fosfatidilinositolo-3-chinasi); fattori trascrizionali; le cicline A, D ed E e le loro chinasi Cdk (chinasi dipendenti dalle cicline); molecole anti- e proapoptotiche (fig. 7). La loro attivazione regola funzioni cellulari differenti, quali l'adesione, la migrazione, la proliferazione, la differenziazione, l'apoptosi e l'espressione genica. Le integrine rappresentano pertanto i più importanti adattatori molecolari attraverso i quali l'ECM può modulare le funzioni cellulari. Molteplici sono le evidenze che dimostrano questo e paradigmatici sono gli effetti delle proteine delle ECM sui cheratinociti. Infatti è stato dimostrato che la proliferazione e la differenziazione dei cheratinociti sono controllate dalle laminine e dalla fibronectina presenti nelle LB dell'epidermide: mentre le laminine, interagendo con l'integrina α6β4, inducono l'attivazione della via di segnalazione di Ras/MAPK e controllano la crescita e la proliferazione dei cheratinociti, la fibronectina, mediante l'integrina α5β1, attivando vie di segnalazione complesse, ne blocca la differenziazione. È questo il meccanismo biologico per cui i cheratinociti in contatto con le LB proliferano e man mano che vi si allontanano possono differenziare.
Le macromolecole dell'ECM: struttura, funzioni e patologie
I glicosamminoglicani
I glicosamminoglicani sono catene di polisaccaridi non ramificate composte da unità di disaccaridi ripetute. Sono dette GAG perché uno dei due residui di zucchero nel disaccaride ripetuto è sempre un amminozucchero (N-acetilglucosammina o N-acetilgalattosammina); il secondo zucchero è di solito un acido uronico (glucuronico o iduronico). In entrambi i residui glicidici sono presenti gruppi solforici o carbossilici che conferiscono ai GAG un'elevata carica negativa. In base ai residui glicidici, al tipo di legame tra i residui e al numero e alla posizione dei gruppi solforici, si distinguono quattro gruppi principali di GAG: (a) l'acido ialuronico; (b) il condroitinsolfato e il dermatansolfato; (c) l'eparansolfato e l'eparina; (d) il cheratansolfato. I GAG tendono ad assumere conformazioni distese, che occupano un volume enorme rispetto alla loro massa, e formano gel anche a concentrazioni molto basse, in quanto a causa dell'elevata densità delle loro cariche negative attraggono una nube di cationi, quali il Na+, che sono osmoticamente attivi, provocando l'afflusso di grandi quantità d'acqua nell'ECM. Infatti, sebbene la quantità di GAG nel tessuto connettivo sia meno del 10% rispetto a quella delle proteine fibrose, la loro capacità di formare gel porosi e idratati fa sì che essi occupino gran parte dell'ECM dei tessuti connettivi, vi permettano la diffusione di molecole idrosolubili e la migrazione cellulare e ne determinino la funzione di supporto meccanico ai vari organi, nonché la resistenza alle forze di compressione. Per mezzo di tale meccanismo, l'ECM della cartilagine che avvolge l'articolazione del ginocchio è in grado di resistere a pressioni di centinaia di atmosfere.
Lo ialuronano (altrimenti detto 'acido ialuronico' o 'ialuronato') è la molecola più semplice di GAG. Per la sua semplicità strutturale, si ritiene che l'acido ialuronico costituisca la prima forma evolutiva di GAG, ma non lo si può considerare rappresentativo della maggioranza dei GAG. Tutti gli altri, infatti, contengono zuccheri solfatati, tendono a comprendere un certo numero di unità disaccaridiche differenti e ordinate in sequenze più complesse, possiedono catene molto più corte, e, unendosi alle proteine mediante legami covalenti, formano i proteoglicani. Per di più, mentre gli altri GAG sono sintetizzati all'interno della cellula e rilasciati per esocitosi, l'acido ialuronico viene direttamente liberato dalla superficie cellulare per mezzo dell'azione di un complesso enzimatico localizzato nella membrana plasmatica. Si ritiene che l'acido ialuronico giochi un ruolo fondamentale nella resistenza dei tessuti e delle articolazioni a forze compressive. Inoltre, durante lo sviluppo embrionale, esso è coinvolto nei complessi cambiamenti morfologici dei tessuti; l'acido ialuronico è anche prodotto in grande quantità durante i processi di riparazione delle ferite ed è un importante componente del fluido delle articolazioni, dove funge da lubrificante.
Fatta eccezione per l'acido ialuronico, tutti gli altri GAG sono legati a proteine a formare i proteoglicani: i più comuni sono l'eparansolfato, il condroitinsolfato, il dermatansolfato e il cheratansolfato. Come nella maggior parte delle glicoproteine, la catena proteica centrale di un proteoglicano viene sintetizzata all'interno della cellula sui ribosomi e traslocata nel lume del reticolo endoplasmatico. Le catene polisaccaridiche vengono successivamente assemblate alla proteina centrale nell'apparato di Golgi, dove molti dei loro residui glicidici sono modificati covalentemente mediante una serie ordinata di reazioni di epimerizzazione e solfatazione. Le reazioni di epimerizzazione modificano la configurazione delle molecole glicidiche; le reazioni di solfatazione, che dipendono dall'attività di un singolo trasportatore di solfati Na+- indipendente, detto DTDST (trasportatore di solfati della displasia diastrofica), aumentano notevolmente la carica negativa dei proteoglicani.
I proteoglicani sono facilmente distinguibili dalle altre glicoproteine in base alla natura, alla quantità e alla disposizione delle loro catene glicidiche laterali: per definizione, almeno una delle catene glicidiche laterali di un proteoglicano deve essere un GAG. Infatti, l'aggrecano, che è un cheratansolfato-condroitinsolfato proteoglicano ed è il maggior componente dell'ECM della cartilagine, possiede più di 100 catene di GAG, e la decorina, che è un dermatansolfato proteoglicano ed è secreta dai fibroblasti, ha una singola catena di GAG. Data la diversa e complessa struttura dei proteoglicani, sarebbe sorprendente che la loro funzione nell'ECM fosse limitata a fornire uno spazio idratato intorno alle cellule. Le catene di GAG dei proteoglicani, infatti, possono formare gel caratterizzati da pori di varie dimensioni e da varie densità di cariche elettriche, e quindi funzionare come setacci selettivi che regolano il traffico delle molecole e delle cellule. Il perlecano, un eparansolfato proteoglicano, svolge proprio questo ruolo nelle LB del glomerulo renale, filtrando le molecole che passano nell'urina dal torrente circolatorio, funzione che il perlecano condivide con un altro eparansolfato proteoglicano, l'agrina, la quale si trova anche nelle giunzioni neuromuscolari.
Si pensa che i proteoglicani giochino inoltre un ruolo essenziale nella trasmissione di segnali chimici tra le cellule. Lo dimostra il fatto che l'FGF (fattore di crescita dei fibroblasti), che stimola una varietà di cellule a proliferare, si lega alle catene dell'eparansolfato e il TGF-β (fattore di crescita trasformante-β), una citochina che agisce ubiquitariamente, si lega alla decorina. Alcuni eparansolfati proteoglicani, quali i sindecani, possono esistere anche come proteine integrali di membrana e agire quindi come modulatori della polarizzazione, della crescita e del differenziamento cellulare: il sindecano può anch'esso legare l'FGF e contemporaneamente associarsi all'actina del citoscheletro, contribuendo a mantenere la morfologia degli strati epiteliali. GAG e proteoglicani si uniscono a formare complessi polimerici nell'ECM: le molecole di aggrecano si assemblano con l'acido ialuronico nello spazio extracellulare in aggregati di notevoli dimensioni. GAG e proteoglicani si associano inoltre a proteine fibrose dell'ECM, come i collageni, e a proteine delle LB, creando strutture estremamente complesse. L'importanza dei GAG è documentata da rare e complesse malattie genetiche dell'uomo, dovute a difetti di sintesi o di accumulo dei GAG; queste ultime sono comunemente conosciute come 'mucopolisaccaridosi' (MPS). Le prime sono principalmente causate da mutazioni del gene che codifica per il DTDST, il trasportatore di solfati Na+-indipendente che controlla non solo la normale solfatazione dei proteoglicani, ma anche la risposta proliferativa dei condrociti, le cellule che principalmente producono i GAG, a fattori di crescita come l'FGF. Esistono quattro forme cliniche di patologie dovute alle alterazioni del DTDST: due di queste (displasia diastrofica e displasia epifisaria multipla tipo 4) sono compatibili con la vita, anche se i soggetti malati sono nani, hanno un aspetto prematuramente invecchiato e difetti generalizzati della pelle, delle articolazioni e delle ossa; le altre due (acondrogenesi tipo IB e atelosteogenesi tipo II) sono invece letali. Recentemente è stato dimostrato che alla base della displasia cartilaginea di Silverman-Handmaker, caratterizzata da nanismo, ci sono mutazioni del gene del perlecano, l'eparansolfato proteoglicano distribuito principalmente nelle LB del glomerulo renale: questa patologia suggerisce pertanto che il perlecano ha un ruolo importante anche nello sviluppo della cartilagine.
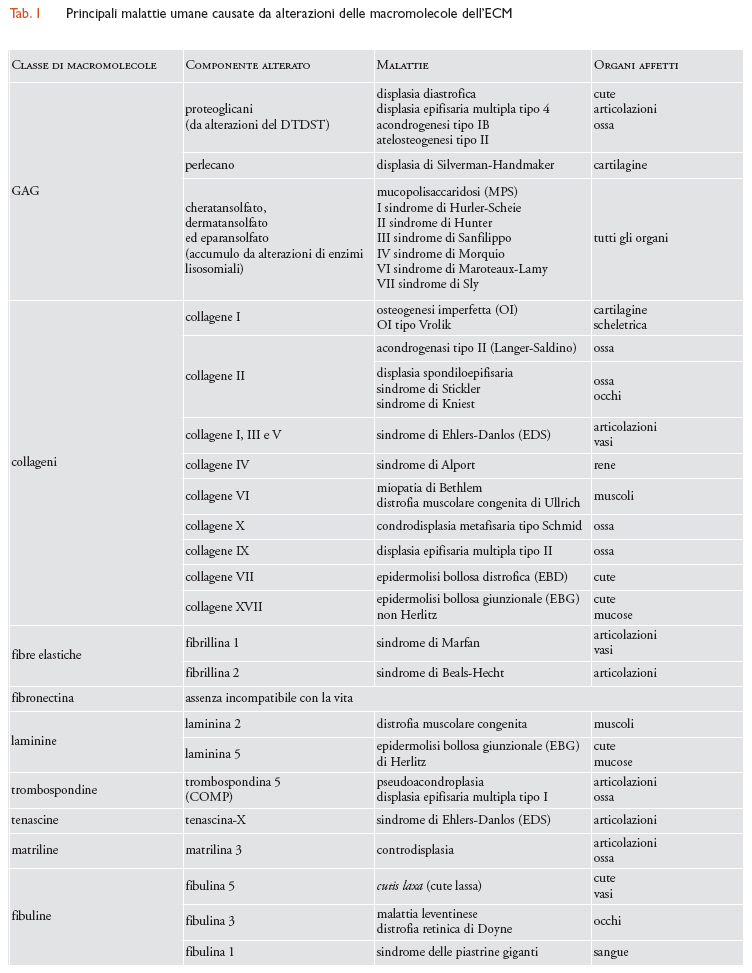
Le mucopolisaccaridosi, al contrario, sono errori congeniti del metabolismo causati da riduzione dell'attività di enzimi lisosomiali, che normalmente degradano i GAG, che pertanto si accumulano in modo eccessivo e dannoso nelle cellule, nei tessuti e negli organi. Le principali sono la MPS I (sindrome di Hurler-Scheie), causata dal deficit dell'enzima lisosomiale α-L-iduronidasi, con accumulo di dermatansolfato ed eparansolfato; la II (sindrome di Hunter), causata dal deficit di iduronato-2-solfatasi e accumulo dei suddetti GAG; la III (sindrome di Sanfilippo), causata da deficit di eparansolfato solfatasi e accumulo di eparansolfato; la IV (sindrome di Morquio), con deficit di N-acetilgalattosamina-6-solfatasi e accumulo di cheratansolfato; la VI (sindrome di Maroteaux-Lamy), con deficit di arilsolfatasi B e deposito di dermatansolfato; la VII (sindrome di Sly), con deficit di β-glucuronidasi e accumulo del suddetto GAG. I pazienti affetti da mucopolisaccaridosi manifestano ritardo fisico e psichico, irrigidimento delle articolazioni e disturbi visivi, uditivi, respiratori, cardiaci e scheletrici. Nella maggior parte dei casi, le mucopolisaccaridosi hanno prognosi severa e sono fatali prima dell'età adulta (tab. 1).
Le proteine fibrose strutturali
I collageni
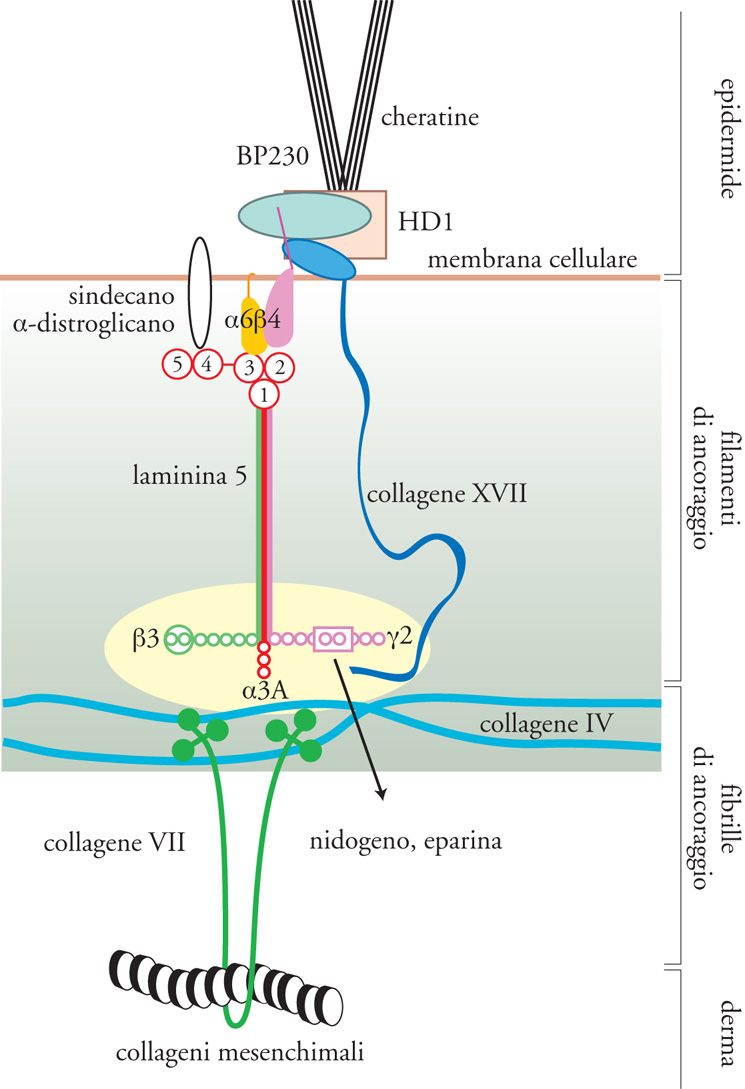
I collageni (Coll) sono la famiglia di proteine fibrose dell'ECM più rappresentate, costituendo il 25% della massa totale delle proteine dei Mammiferi. Sono i collageni dell'ECM a fornire l'intelaiatura strutturale ai tessuti e contrariamente ai GAG, che resistono alle forze compressive, i collageni resistono alla trazione. Esistono almeno 27 tipi distinti di collagene. I tipi I, II, III, V e XI costituiscono i collageni interstiziali o fibrillari, sono i più abbondanti e sono detti 'collageni classici'. Collageni interstiziali sono considerati anche i tipi XVI, XIX, XX, XXI e XXII, recentemente identificati. I tipi IV, VI e VII sono non fibrillari o 'amorfi' e sono presenti nel tessuto interstiziale e nelle LB. Mentre il tipo VI è ubiquitario, i tipi IV e VII sono chiamati 'collageni che formano reti': le molecole di tipo IV si organizzano in una rete che costituisce la maggior parte delle LB, mentre le molecole di tipo VII formano dimeri che si organizzano in strutture specializzate dette 'fibrille di ancoraggio', che fanno sì che le LB degli epiteli multistratificati si uniscano al tessuto connettivo sottostante. A questo gruppo di collageni appartengono anche i tipi VIII, XV e XVIII, definiti 'multiplexine', che sono stati rinvenuti nelle LB di epiteli ed endoteli di vari tessuti. Recentemente è stato dimostrato anche che il collagene XVIII, detto anche 'endostatina', si lega al GAG eparansolfato. I tipi IX e XII sono detti 'collageni associati alle fibrille' in quanto si trovano alla superficie delle fibrille di altri tipi di collagene, legandole tra di loro e ad altre componenti dell'ECM, come il GAG condroitinsolfato. Recentemente è stato identificato un gruppo di collageni associati alle membrane delle cellule: i tipi XIII, XVII, XXIII e XXV. Di questi, i più importanti sono il tipo XVII, conosciuto anche come BP180, l'antigene del peso molecolare di 180 kDa del pemfigoide bolloso, espresso a livello delle LB delle giunzioni dermo-epidermiche (fig. 8), e il tipo XXV, definito anche CLAC-P (precursore del componente simile al collagene delle placche amiloidee dell'Alzheimer), coinvolto nella degenerazione neuronale del morbo di Alzheimer. Infine, è stato isolato un nuovo tipo di collagene, il XXVI, che presenta caratteristiche simili all'emilina, una proteina associata alle fibre elastiche. Molti di questi tipi di collagene si legano ai recettori integrinici: paradigmatico è il collagene I, che, mediante la sequenza DGEA, si lega all'integrina α2β1.
Nonostante la loro varietà e complessità, la caratteristica peculiare di una tipica molecola di collagene, come quella dei collageni classici, è la struttura a tripla elica lunga e rigida, costituita da tre catene polipeptidiche dette 'catene α', avvolte l'una intorno all'altra a formare un'elica superavvolta avente l'aspetto di una fune. Esistono 42 tipi distinti di catene α, indicate progressivamente come α1, α2, α3, e così di seguito, e codificate da altrettanti e corrispondenti geni. Tutti i 27 tipi di collageni sono costituiti da diverse combinazioni delle varie catene α: l'attuale nomenclatura molecolare dei collageni prevede infatti che oltre al tipo (I, II, III, ecc.) si indichi anche il tipo di catene α da cui è costituito. I collageni sono estremamente ricchi in prolina e glicina, amminoacidi entrambi importanti nella formazione della tripla elica. La prolina, grazie alla sua struttura ad anello, stabilizza la conformazione a elica in ciascuna catena α, mentre la glicina fa sì che le tre catene α disposte a elica siano strettamente compattate per formare la superelica finale del collagene. Le singole catene polipeptidiche di collagene vengono sintetizzate sotto forma di precursori più grandi, detti 'pro-catene α'. In questa fase le catene α sono soggette a diverse modificazioni enzimatiche, tra cui l'idrossilazione dei residui di prolina e di lisina, evento per cui è necessaria la vitamina C o acido ascorbico. Ogni pro-catena α si combina poi con altre due formando una molecola a tripla elica, nota come 'procollagene', che contiene propeptidi che vengono eliminati durante o immediatamente dopo la secrezione, a opera delle peptidasi. Le molecole di collagene a questo punto si riuniscono a formare fibrille, che si rafforzano notevolmente perché si creano legami trasversali molto stabili tra i residui di lisina delle molecole di collagene stesso.
Le fibrille possiedono diametro variabile e sono organizzate in modo diverso a seconda dei tessuti, dove sono le cellule stesse a determinarne la dimensione e la disposizione. Nella cute dei Mammiferi si avvolgono a costituire una trama che assomiglia a un intreccio di vimini, in modo da resistere alle sollecitazioni applicate in più direzioni. Nei tendini le fibrille di collagene sono organizzate in fasci paralleli allineati lungo l'asse principale della tensione esercitata sul tendine. Sia nel tessuto osseo di un organismo adulto, sia nella cornea le fibrille di collagene sono disposte ordinatamente in strati. Dato l'elevato numero di passaggi enzimatici coinvolti nella formazione di una fibrilla di collagene, non sorprende il fatto che esistano molte malattie dovute a difetti di formazione delle fibrille. Nello scorbuto, una malattia comune nei marinai fino al secolo scorso e dovuta a una carenza di vitamina C, le pro-catene α sintetizzate sono alterate, non possono formare una tripla elica stabile e sono immediatamente degradate nella cellula. Il collagene, che viene fisiologicamente degradato nei tessuti, non può essere più sostituito: i vasi sanguigni diventano estremamente fragili e i denti cominciano a muoversi negli alveoli.
Più di 1300 mutazioni sono state finora identificate in ben 23 dei 42 geni che codificano per le corrispondenti catene α che costituiscono i 27 tipi di collageni noti. Molteplici, pertanto, sono le alterazioni geneticamente trasmesse dei collageni, causa di differenti e complesse patologie. Tra queste emerge per gravità l'osteogenesi imperfetta (OI), che definisce un gruppo di quattro sindromi (tipo I, II, III e IV) clinicamente eterogenee, il cui comun denominatore è uno stato di abnorme fragilità ossea dovuta al ridotto contenuto di collagene I e a cui si associano tratti fenotipici fortemente caratteristici come le sclere blu, ma anche compromissione della funzione uditiva e anomalie dell'apparato cardiovascolare. La variante nota come OI tipo II (o tipo Vrolik) è letale, in quanto nessun individuo affetto sopravvive oltre i primi giorni dopo la nascita, a causa delle numerose fratture che si producono in utero per effetto delle contrazioni muscolari durante i movimenti fetali o durante il parto.
Complesso è anche il gruppo di patologie definite 'condrogenesi imperfetta', caratterizzate dalla riduzione del contenuto di collagene II nella matrice cartilaginea e pertanto dall'abnorme costituzione e sviluppo delle cartilagini scheletriche. Tra le forme più gravi, si ricorda l'acondrogenesi tipo II (Langer-Saldino), una malattia letale perinatale, caratterizzata dalla pressoché totale assenza di ossificazione nel rachide; tra le meno gravi, la displasia spondiloepifisaria congenita è riconosciuta per l'evidente accorciamento degli arti e anomalie oculari fino alla cecità, che divengono il dato clinico più appariscente nella sindrome di Stickler e nella sindrome di Kniest. Recentemente si è osservato che anomalie del collagene II, verosimilmente risultanti da mutazioni misconosciute, possono associarsi a forme precoci di artrosi, e perfino ad alcune forme di miopia progressiva, cataratta e glaucoma. Ad alterazioni dei collageni I, III e V sono state attribuite alcune forme di sindrome di Ehlers-Danlos (EDS), caratterizzata dalla presenza di vasi sanguigni fragili e articolazioni particolarmente mobili. La forma cifoscoliotica, con artroclasia (rottura di un'articolazione) e con dermatosparassi (cute fragile e che si strappa facilmente) è dovuta a difetti nella corretta maturazione del collagene I; la forma vascolare, la più grave, dove la pelle è sottile e trasparente e i grandi vasi sanguigni possono andare incontro a rotture spontanee, è dovuta ad alterazioni del collagene III; la forma classica, con articolazioni ipermobili e tessuti abnormemente elastici e fragili che causano ecchimosi, ernie e prolassi, è causata da alterazioni del collagene V.
Il difetto nella sintesi del collagene IV, costituente fondamentale della membrana basale del glomerulo renale, è alla base della sindrome di Alport, che conduce a una progressiva nefropatia. Recentemente si è dimostrato che mutazioni dei geni del collagene VI sono responsabili della miopatia di Bethlem, caratterizzata da atrofia e debolezza dei muscoli del tronco e degli arti, e della distrofia muscolare congenita di Ullrich, che si manifesta con debolezza generalizzata e progressiva insufficienza respiratoria. Mutazioni del gene che codifica per il collagene X causano la condrodisplasia metafisaria tipo Schmid, che comporta bassa statura e incurvamento delle ossa lunghe degli arti; mutazioni del gene che codifica per il collagene IX danno origine alla displasia epifisaria multipla tipo II, caratterizzata anch'essa da nanismo e arti corti. Infine, alterazioni dei collageni possono causare varie forme di epidermolisi bollosa (EB), un gruppo eterogeneo di malattie caratterizzate da fragilità della cute e delle mucose e dalla formazione di bolle. In particolare, mutazioni nel gene che codifica per il collagene VII sono alla base della epidermolisi bollosa distrofica o dermolitica (EBD), in cui la localizzazione delle lesioni bollose è prevalentemente cutanea, con lenta guarigione e cicatrici retraenti, mentre alterazioni del collagene XVII causano una forma di epidermolisi bollosa giunzionale (EBG), quella non Herlitz, con interessamento non solo cutaneo, ma anche delle mucose orale, nasale, oculare e genitale (tab. 1).
L'elastina
La funzionalità dei tessuti che costituiscono i vasi sanguigni, la cute, i polmoni, l'utero e l'occhio richiede elasticità, che è assicurata dalla presenza nella loro ECM di una fitta rete di fibre elastiche. La componente principale delle fibre elastiche è l'elastina, una proteina idrofobica. Le molecole di elastina vengono secrete nello spazio extracellulare, si aggregano in fibre elastiche e stabiliscono un gran numero di legami trasversali l'una con l'altra. Mutazioni del gene dell'elastina sono alla base di patologie come la stenosi sopravalvolare e la cutis laxa (cute lassa), caratterizzata, quest'ultima, dalla presenza di pliche cutanee pendule, arterie tortuose ed enfisema. Le fibre elastiche, tuttavia, non sono composte solamente da elastina. Infatti, il nucleo centrale dell'elastina è coperto da una guaina di microfibrille.
Si pensa che le microfibrille giochino un ruolo importante nell'aggregazione delle fibre elastiche. Esse compaiono prima dell'elastina nei tessuti in via di sviluppo e costituiscono lo scheletro su cui le molecole di elastina secrete vengono depositate. Le microfibrille sono composte da diverse glicoproteine, incluse le fibrilline 1, 2 e 3, che sembrano essere essenziali per l'integrità delle fibre elastiche, e da due nuove classi di proteine: le MAGP1 e 2 (glicoproteine 1 e 2 associate alle microfibrille) e le emiline che legano le fibre elastiche alle superfici cellulari. Mutazioni nel gene della fibrillina 1 causano la sindrome di Marfan, una malattia genetica che colpisce i tessuti connettivi ricchi in fibre elastiche; negli individui ammalati più gravemente, oltre a difetti scheletrici e oculari, l'aorta, le cui pareti normalmente sono molto ricche di elastina, è addirittura suscettibile di rottura, come si verifica nell'aneurisma dissecante dell'aorta. Mutazioni nel gene della fibrillina 2 causano l'aracnodattilia contratturale congenita, o sindrome di Beals-Hecht, una rara malattia caratterizzata da arti ad aspetto marfanoide e dita sproporzionatamente lunghe.
Le proteine fibrose adesive
La fibronectina
La fibronectina (FN) è la prima delle proteine adesive dell'ECM a essere stata caratterizzata. Queste proteine hanno il compito sia di organizzare l'ECM sia di permettere alle cellule di connettersi con essa. La fibronectina si lega a diversi altri componenti dell'ECM, tra cui i collageni, in modo particolare al collagene di tipo III, e ai proteoglicani tramite domini specifici, e alle cellule tramite i recettori integrinici. La fibronectina è una grossa glicoproteina composta da due subunità, unite da due legami disolfuro all'estremità carbossiterminale della molecola. Ciascuna subunità è ripiegata in una serie di domini funzionali, che consistono di moduli ripetuti. Il tipo principale di modulo, detto repeat di tipo III della fibronectina, è presente almeno 15 volte in ciascuna subunità. In alcuni di questi moduli si trovano le sequenze amminoacidiche specifiche RGD e EILDV, che giocano un ruolo importante nella sua interazione con molti recettori appartenenti alla famiglia delle integrine β1 e β3. Esistono circa 20 forme multiple (isoforme) di fibronectina, che si organizzano sulla superficie delle cellule e vengono depositate nell'ECM o nelle membrane basali come filamenti di fibronectina altamente insolubili. La fibronectina è prodotta da fibroblasti, cellule endoteliali e leucociti. Si ritiene che la fibronectina aumenti la sensibilità di alcuni tipi cellulari (come le cellule endoteliali dei capillari) agli effetti proliferativi dei fattori di crescita e sia coinvolta direttamente nell'adesione, nello spreading (il processo col quale la cellula si appiattisce sul substrato, aderendovi attivamente), nella migrazione, nella proliferazione, nella differenziazione e nella modulazione dell'apoptosi delle cellule dei tessuti. È presente anche un'isoforma plasmatica della fibronectina, che è solubile e circola nel sangue e in altri fluidi corporei, dove si pensa che faciliti la coagulazione sanguigna, la riparazione delle ferite e la fagocitosi.
La fibronectina è anche implicata nella trasformazione e progressione, nonché nella metastatizzazione dei tumori. Le cellule tumorali presentano spesso alterazioni quantitative e qualitative della sintesi di fibronectina: la presenza di specifiche isoforme di fibronectina nelle lesioni tumorali può esssere utilizzata come un marcatore diagnostico. Variazioni dell'espressione e della funzione dei recettori integrinici specifici per essa, quali l'α5β1, sono anch'esse state associate con le differenti fasi di sviluppo di un tumore. Infatti, la perdita dell'espressione di questa integrina correla con un aumento del potere trasformante, invasivo e metastatico tumorale, che può essere controllato utilizzando peptidi contenenti la sequenza RGD che bloccano l'interazione dell'α5β1 con la fibronectina. L'importanza della fibronectina va al di là delle funzioni sopraelencate: topi che hanno subito una mutazione del gene della fibronectina, muoiono in stadi precoci di sviluppo e presentano difetti morfologici multipli a carico del cuore, dei vasi sanguigni, del tubo neurale e delle membrane extraembrionali. Nell'uomo non sono state riscontrate alterazioni quantitative e qualitative della fibronectina al di là di quelle rilevate nei tumori; è stato descritto che la sua assenza è in ogni caso incompatibile con la vita.
Le laminine
Le laminine (LM) sono le glicoproteine più abbondanti nell'ECM organizzata in quelle strutture altamente specializzate che sono le LB. La prima laminina scoperta è stata isolata dall'ECM del sarcoma di Engelbreth-Holm-Swarm (EHS) e inizialmente sembrava essere costituita da due catene, tenute insieme da ponti disolfuro. Studi successivi hanno dimostrato una grande variabilità di questa proteina, dovuta alla presenza di tre catene geneticamente distinte ma omologhe: le catene α, β e γ. Si è visto che la laminina derivata dal tumore EHS è solo un membro di una grande famiglia di proteine costituita dalle diverse combinazioni di queste catene di cui esistono molteplici varianti. Infatti, sono state individuate cinque catene α (da α1 ad α5), tre differenti catene β (da β1 a β3) e tre differenti catene γ (da γ1 a γ3), le cui combinazioni potrebbero ipoteticamente portare alla formazione di 45 isoforme differenti di laminine. Tuttavia esistono limitazioni a queste combinazioni: la catena γ2 si associa solo con le catene α3 e β3, formando la laminina 5. Finora è stata dimostrata la presenza di 12 isoforme che possono differire tra loro per una, due o tre catene α, β e γ. Data questa grande variabilità, è stata adottata una nuova nomenclatura che definisce la composizione specifica di ognuna delle 12 isoforme caratterizzate. La laminina 1 o EHS-laminina è α1β1γ1; la laminina 2 o merosina è α2β1γ1; la laminina 3 o S-laminina è α1β2γ1; la laminina 4 o merosina/S-laminina è α2β2γ1; la laminina 5 o kalinina/niceina è α3β3γ2; la laminina 6 o K-laminina è α3β1γ1; la laminina 7 o KS-laminina è α3β2γ1; la laminina 8 è α4β1γ1; la laminina 9 è α4β2γ1; la laminina 10 è α5β1γ1; la laminina 11 è α5β2γ1; infine l'ultima laminina, scoperta nel 1999, è la laminina 12, la cui composizione è α2β1γ3.
Undici delle dodici forme note della laminina mostrano un'espressione ristretta alle LB. Per semplicità possiamo analizzare la loro distribuzione nelle LB basandoci su quella delle catene α, β e γ che le costituiscono. Le laminine che contengono le catene α si dividono in quelle α1, α3 e α5, che si trovano nelle LB di tutti gli epiteli, in quelle α2, che costituiscono le LB di tessuti mesenchimali, come i muscoli e il cuore, e in quelle α4, che sono componenti principali delle LB dei grossi vasi come l'aorta, ma anche dei tessuti adiposi. Le laminine che contengono le catene β1 sono ampiamente diffuse in tutte le LB, mentre quelle costituite dalle β2 sono presenti nel mesangio dei glomeruli renali e nelle LB che sostengono gli alveoli polmonari. L'unica laminina costituita dalle catene β3 e γ2 è la laminina 5, che è il costituente principale delle LB degli epiteli stratificati, come l'epidermide (fig. 8). Le laminine che contengono le catene γ1 sono le più ubiquitarie e le loro alterazioni sono embriologicamente letali. Le catene γ3 sono alla base della formazione della laminina 12: questa è l'unica laminina che non è presente in specifiche LB, ma si trova nell'intestino, utero, testicolo, ovaio, placenta, polmoni e sistema nervoso.
Tutte le diverse isoforme di laminina hanno comunque in comune l'organizzazione tridimensionale: ogni laminina, infatti, è una molecola cruciforme asimmetrica con un braccio lungo e due corti, e tutte le laminine presentano nella loro struttura domini multipli, responsabili delle loro complesse interazioni con gli altri costituenti delle LB e con recettori cellulari. Infatti le laminine attraversano le LB, legandosi da un lato al collagene di tipo IV e all'eparansolfato e, dall'altro, a recettori cellulari specifici di tipo integrinico, quali le integrine α1β1, α2β1, α3β1, α6β1, α7β1 e α6β4, e di tipo non integrinico. Il complesso distrofina-destroglicano-sarcoglicani funziona come recettore non integrinico e connette le laminine al citoscheletro delle cellule muscolari. Nel cuore adulto normale, le laminine sono localizzate a ridosso della membrana plasmatica dei cardiomiociti, dove si connettono con il collagene di tipo IV, e in misura minore nei fibroblasti cardiaci, dove legano i collageni di tipo I e III. Le laminine sono presenti anche nel tessuto muscolare a livello dei tubuli T e sono molto abbondanti in aree di specializzazione morfologica come le bande Z del sarcomero, mediando la connessione tra i fasci di collageni e la membrana del sarcolemma. Si ritiene che le varie isoforme delle laminine possano mediare l'adesione delle cellule a substrati dei tessuti connettivi e modulare la crescita, la sopravvivenza, la morfologia, il differenziamento e la motilità di diversi tipi cellulari. Molte di queste funzioni sono mediate dall'interazione delle laminine con i recettori integrinici e in particolare con l'integrina α6β4.
Complesse sono le malattie genetiche riconducibili a deficit qualitativi o quantitativi delle varie isoforme della laminina. Mutazioni geneticamente trasmesse delle catene α2, che sono presenti principalmente nella laminina 2 (o merosina), sono la causa della distrofia muscolare congenita, la più frequente tra la popolazione caucasica e definita anche 'tipo classico' per il coinvolgimento esclusivamente muscolare. La merosina è la laminina più rappresentata nelle LB delle fibre muscolari, dove lega l'α-distroglicano, il quale è connesso mediante il β-distroglicano alla distrofina del citoscheletro della cellula muscolare. L'assenza di merosina interrompe questo legame e porta a degenerazione del muscolo. Inoltre, la merosina è importante per la mielinizzazione e quindi la sua assenza provoca una neuropatia demielinizzante. I soggetti che presentano difetto completo di merosina hanno di solito un quadro clinico piuttosto severo e non acquisiscono la deambulazione. Anche il sistema nervoso centrale è coinvolto, sia pure in forma subclinica: le capacità cognitive sono abitualmente del tutto adeguate e solo in alcuni casi si presentano lievi anomalie della vista. Mutazioni nei geni che codificano per le tre catene della laminina 5 (o kalinina/niceina) causano l'epidermolisi bollosa giunzionale (EBG) di Herlitz, a esordio connatale, con lesioni bollose ed erosioni localizzate non solo all'intera superficie cutanea, ma anche a tutte le mucose degli apparati digerente, respiratorio e genito-urinario. La prognosi è infausta, di solito entro il primo anno di vita, per complicanze infettive o secondarie al coinvolgimento mucoso (tab. 1).
Le entactine o nidogeni
Tutte le LB, oltre a contenere laminine, collagene di tipo IV e di tipo XVIII, i proteoglicani perlecano e argina, sono costituite anche dalla glicoproteina definita 'entactina-1/nidogeno-1' (fig. 8). La scoperta che l'inibizione delle interazioni tra questa proteina e le laminine perturbano l'organogenesi e la formazione delle LB, ha evidenziato la sua importanza nell'organizzazione delle LB, dove essa forma complessi con tutti i componenti sopra descritti. Sembra inoltre che l'entactina-1/nidogeno-1 sia in grado di promuovere l'adesione cellulare, la chemiotassi dei neutrofili, la crescita del trofoblasto e l'angiogenesi. È stato recentemente dimostrato che esiste una seconda isoforma denominata 'entactina-2/nidogeno-2/osteonidogeno', che presenta con la prima un'elevata omologia amminoacidica e strutturale, nonché una distribuzione cellulare sovrapponibile, sebbene sia maggiormente espressa nel cuore e nel muscolo scheletrico e si differenzi per la capacità di legare differenti substrati e recettori integrinici.
La vitronectina
La vitronectina (VN) è una glicoproteina sintetizzata principalmente dal fegato, che circola nel plasma dove modula l'attivazione del sistema del complemento e si deposita nei tessuti dove diviene un componente dell'ECM e interagisce con l'elastina, i glicosamminoglicani e i collageni. Analogamente alla fibronectina, essa è costituita da molteplici domini. Tra questi, la regione di connessione, che contiene la sequenza amminoacidica specifica RGD, interagisce con numerose integrine della famiglia della β1 e della β3, mentre il dominio somatomedina B lega il PAI-1 (inibitore -1 dell'attivatore del plasminogeno) e l'uPAR (recettore dell'attivatore del plasminogeno di tipo urochinasi). Queste interazioni sono alla base di due nuove e complesse funzioni attribuite alla vitronectina: modulazione dell'angiogenesi e dell'invasione e metastatizzazione delle cellule tumorali. Il legame delle integrine β3 alla vitronectina è importante per la formazione di nuovi vasi; l'attivazione del sistema del plasminogeno, mediata dall'uPA (attivatore del plasminogeno di tipo urochinasi) rilasciato dalle cellule tumorali e modulata dalla vitronectina, porta alla formazione di plasmina che catalizza la degradazione delle LB e dell'ECM, favorendo in tal modo i processi di invasione e diffusione metastatica.
Le proteine matricellulari
La SPARC/osteonectina
La SPARC, conosciuta anche come osteonectina o BM-40 (proteina del peso molecolare di 40 kDa della matrice dell'osso), è il prototipo delle proteine matricellulari. È una proteina modulare che presenta tre domini strutturali con cui lega il calcio, proteine strutturali dell'ECM, quali i collageni e la vitronectina, e fattori di crescita come il PDGF (fattore di crescita derivato dalle piastrine) e il VEGF (fattore di crescita dell'endotelio vascolare). La SPARC è stata inizialmente descritta come il costituente principale dell'osso, dove, in quanto dotata di alta affinità per il calcio sia come ione libero che associato in complessi di tipo cristallino, si ritiene che essa agisca come elemento di nucleazione dei cristalli di minerali: la SPARC sarebbe in grado di concentrare il calcio nelle sue adiacenze creando così le condizioni per avviare la precipitazione del fosfato di calcio. L'osservazione che osteoblasti isolati da pazienti affetti da osteogenesi imperfetta producono basse quantità di SPARC, testimonia l'importanza del suo ruolo biologico nel tessuto osseo. La SPARC è inoltre espressa durante l'embriogenesi e in tessuti sedi di processi fisiologici, quali il rimodellamento, e patologici, quali tumori, artriti, diabete, dove potrebbe, regolando l'adesione e la proliferazione cellulare, controllare l'angiogenesi e la sintesi e la degradazione dell'ECM.
Le trombospondine
Le trombospondine (TSP) sono una famiglia di glicoproteine multimodulari leganti il calcio. Il prototipo di questa famiglia è la trombospondina 1 (TSP-1), che lega la fibronectina e regola l'aggregazione piastrinica, la risposta infiammatoria e l'angiogenesi durante i processi di riparazione e la crescita tumorale. Condivide molte di queste attività biologiche con la trombospondina 2 (TSP-2), che è anche coinvolta nell'organizzazione dell'ECM. Delle altre trombospondine finora caratterizzate, è accertato che le trombospondine 3, 4 e 5 legano con diversa specificità i collageni fibrillari di tipo I, II, e IX, le laminine e la fibronectina. La trombospondina 5, definita anche COMP (proteina oligomerica della matrice cartilaginea), in quanto principalmente espressa nella cartilagine, è stata recentemente implicata nell'adesione e differenziazione dei condrociti. Il ruolo biologico delle trombospondine si amplia ulteriormente se si considera che esse possono influenzare le funzioni di vari tipi cellulari sia direttamente, modulando l'organizzazione del citoscheletro, la migrazione e la proliferazione, sia indirettamente, regolando l'attivazione o l'inibizione di proteasi, fattori di crescita e citochine, quali il TGF-β. Recentemente si è osservato che due forme di displasia ossea, la pseudoacondroplasia e la displasia epifisaria multipla tipo I, sono causate da mutazioni del gene che codifica per la trombospondina 5 o COMP. La prima è una delle displasie scheletriche in assoluto più comuni ed è una forma di nanismo ad arti corti, che si manifesta verso il secondo anno di età; la seconda è caratterizzata da minore riduzione della statura, dalla restrizione dei movimenti articolari e dalla frequente necrosi della testa del femore. Ne esistono due varianti cliniche, dette rispettivamento 'tipo Fairbank' e 'tipo Ribbing', la prima più compiutamente espressa della seconda (tab. 1).
L'osteopontina
L'osteopontina (OPN) è da sempre considerata come la proteina ossea strutturale che connette le cellule ossee, gli osteoblasti, all'ECM. Fa parte della famiglia SIBLING (glicoproteine N-legate con piccole regioni di legame per le integrine), un gruppo di proteine ossee e dentali che presentano, come la fibronectina, delle sequenze RGD. È una proteina intracellulare, ma viene anche secreta ed è in grado di legare minerali come l'idrossiapatite, fattori del sistema del complemento come il fattore H, e recettori cellulari come l'integrina αvβ3. Le sue funzioni sono pleiotropiche: l'osteopontina non solo inibisce la mineralizzazione nelle ossa e la precipitazione dei soluti nelle urine, ma funziona anche come potente attivatore della migrazione dei leucociti. È proprio grazie a quest'attività proinfiammatoria che l'osteopontina è oggi implicata nella modulazione della risposta immunitaria a infezioni da micobatteri o da herpes virus e nei tumori prostatici e mammari. Recentemente è stato suggerito il suo coinvolgimento anche in malattie autoimmunitarie come la sclerosi multipla.
Le tenascine
Le tenascine (TN) sono una famiglia di grandi proteine multimeriche dell'ECM. Esse hanno una distribuzione tessutale molto ristretta e sono più abbondanti nell'ECM dei tessuti embrionali. Si conoscono quattro tenascine: la tenascina C o antigene miotendineo o citotactina; la tenascina R o restrictina o gianusina; la tenascina X e la tenascina N. Tutte presentano una struttura simile, costituita da sei catene polipeptidiche identiche o simili unite da ponti disolfuro che si irradiano da un centro come i raggi di una ruota. Analogamente alla fibronectina, ciascuna delle catene polipeptidiche è composta da molti tipi di corte sequenze amminoacidiche ripetute più volte ed è ripiegata in numerosi domini funzionalmente distinti. Di questi, alcuni si legano al proteoglicano transmembrana sindecano, altri si legano alla fibronectina. Le tenascine possono influenzare la forma, l'adesione, la migrazione e la crescita cellulare. Anche se si è dimostrato che topi che hanno subito mutazioni dei geni delle tenascine presentano malformazioni nervose, cutanee e dei processi rigenerativi, a tutt'oggi non si conoscono patologie umane causate da alterazioni delle tenascine. Tuttavia alcuni tumori presentano un'aumentata espressione della tenascina C, che viene pertanto utilizzata come un marcatore diagnostico. Sebbene molte forme di sindrome di Ehlers-Danlos siano dovute ad alterazioni dei collageni, è ampiamente dimostrato che la forma recessiva di questa patologia è causata da un difetto della tenascina-X: i pazienti affetti presentano ipermobilità delle articolazioni, pelle iperelastica ed estrema facilità alle ecchimosi (tab. 1).
Le matriline
Le matriline costituiscono una nuovissima famiglia di quattro proteine multimodulari e multifunzionali dell'ECM, caratterizzate dalla presenza di domini simili al VWA (fattore A di von Willebrand). Sebbene siano principalmente espresse nella cartilagine, esse partecipano anche alla formazione delle componenti fibrillari e filamentose dell'ECM e sono solitamente associate ai collageni. Sembra che esse siano coinvolte nelle interazioni tra le fibrille di collagene e i proteoglicani. La matrilina 1, definita anche CMP (proteina della matrice della cartilagine), è il prototipo di questa famiglia: essa lega stabilmente i proteoglicani aggrecano e decorina, ma anche un altro componente della cartilagine, la trombospondina 5 o COMP. Le altre matriline, le matriline 2, 3 e 4, sono strutturalmente simili alla 1, ma variano per dimensioni e localizzazione. Recentemente si è osservato che mutazioni genetiche della matrilina 3 causano osteoartriti delle mani e differenti forme di condrodisplasia, tra cui una variante della displasia epifisaria multipla, caratterizzata da un'ossificazione ritardata e irregolare delle epifisi ossee e comparsa precoce di osteoartriti (tab. 1).
Le fibuline
Di recentissima scoperta sono anche le fibuline, una famiglia di sei glicoproteine il cui nome deriva dal latino fibula, che significa fibbia, e che sono caratterizzate dalla presenza di un dominio di 120÷140 amminoacidi che le contraddistingue. La fibulina 6 è anche nota come 'emicentina 1'. Studi biochimici hanno successivamente indicato che sia la 1 che la 2, le più voluminose e rappresentate, si associano con le fibre elastiche, le microfibrille di fibronectina, gli aggregati di proteoglicani e i componenti delle LB, quali le laminine, le entactine o nidogeni e il collagene XVIII. Esse sono chiaramente espresse durante lo sviluppo embrionale nell'apparato cardiovascolare e nei siti di trasformazione epiteliale-mesenchimale, mentre nell'adulto la loro distribuzione tessutale varia e diventano maggiormente espresse nella cute, nel polmone, nei vasi e nell'occhio. Nuovissimi studi genetici e molecolari hanno associato alterazioni di alcuni dei geni che codificano per le fibuline con molteplici malattie ereditarie che colpiscono proprio questi organi. Infatti, alterazioni del gene della fibulina 5 sono state associate con alcuni casi di cutis laxa. Due malattie, la malattia leventinese e la distrofia retinica di Doyne, in cui si verifica una progressiva degenerazione della macula, la regione della retina posteriore con alta densità di recettori per gli stimoli visivi, sono causate da alterazioni a carico di geni che mappano in prossimità del gene della fibulina 3. Le suddette patologie sono caratterizzate dall'accumulo nella retina di depositi extracellulari (noti come 'druse'), che conducono a una progressiva e invalidante perdita della capacità visiva. Alcune forme di distrofia retinica, come la degenerazione maculare legata all'età, sono state attribuite anche ad alterazioni della fibulina 6 o emicentina 1 e alla diminuita espressione della fibulina 1. Quest'ultima può inoltre essere alla base di malformazioni congenite delle mani e della sindrome delle piastrine giganti, con macrotrombocitopenia e corpi inclusi nei neutrofili. Infine, si è osservato che la fibulina 1 è anche implicata nel controllo della progressione di alcuni tumori come i carcinomi della mammella (tab. 1).
Le emiline
Le emiline ‒ proteine (INE) localizzate (L) nell'interfaccia (I) delle microfibrille (M) di elastina (E) ‒ sono una famiglia di glicoproteine dell'ECM, con caratteristiche simili al collagene XXVI e identificate dalla presenza di una regione ricca di cisteine, denominata 'dominio EMI'. L'emilina-1, la prima proteina di questa famiglia a essere stata scoperta, è prodotta da cellule che sintetizzano e depositano l'elastina nell'ECM, quali le cellule muscolari liscie, i fibroplasti e le cellule endoteliali. Essa è stata inizialmente isolata dall'aorta e successivamente identificata in molteplici tessuti connettivi in associazione con le fibre elastiche e in particolare nell'interfaccia tra il nucleo centrale dell'elastina e le microfibrille circostanti. Simili all'emilina-1 sono anche le altre proteine di questa famiglia, l'emilina-2, l'emilina-3, e la multimerina-2, queste ultime entrambe presenti anche sulle piastrine e sulle cellule endoteliali. Considerando che l'emilina-1 è coinvolta nell'adesione cellulare e lega l'elastina e la fibulina-5, è stato ipotizzato che essa regoli l'elastogenesi e il mantenimento delle strutture vascolari.
Da tutto questo emerge la complessità molecolare, strutturale e funzionale dell'ECM. Non deve sorprendere, quindi, che la sua caratterizzazione continui a essere uno dei maggiori obbiettivi dell'attività di ricerca della comunità scientifica internazionale, se si pensa che senza i glicosamminoglicani e i proteoglicani un essere umano sarebbe schiacciato dalla forze compressive, senza i collageni sarebbe ridotto a un ammasso di cellule connesse da qualche neurone, senza le laminine la sua pelle si staccherebbe e senza la fibronectina, infine, esso non potrebbe nascere. La sempre più approfondita conoscenza della biologia delle macromolecole dell'ECM permette di disegnare farmaci specifici atti a regolarne l'espressione e la funzione. Le innovative tecniche di ingegneria genetica consentono di riprodurle per poter modulare la crescita, la sopravvivenza, il differenziamento, la morfologia e la motilità delle cellule dei tessuti e per la cura e il trattamento di malattie in cui risultano alterate o deficitarie.
Bibliografia
Adams 2001: Adams, Josephine C., Thrombospondins: multifunctional regulators of cell interactions, "Annual review of cell and developmental biology", 17, 2001, pp. 25-51.
Brekken, Sage 2000: Brekken, Rolf A. - Sage, E. Helene, SPARC, a matricellular protein: at the crossroads of cell-matrix, "Matrix biology", 19, 2000, pp. 569-80 (rist. con correzioni: "Matrix biology", 19, 2001, pp. 816-827).
Chabas 2005: Chabas, Dorothée, Osteopontin, a multi-faceted molecule, "Médecine sciences", 21, 2005, pp. 832-838.
Chiquet-Ehrismann 2004: Chiquet-Ehrismann, Ruth, Tenascins, "International journal of biochemistry and cell biology", 36, 2004, pp. 986-990.
Chu, Tsuda 2004: Chu, Mon-Li - Tsuda, Teruko, Fibulins in development and heritable disease, "Birth defects research. Part C. Embryo today", 72, 2004, pp. 25-36.
Colombatti 2000: Colombatti, Alfonso e altri, The EMILIN protein family, "Matrix biology", 19, 2000, pp. 289-301.
Defilippi 1997: Defilippi, Paola e altri, Signal transduction by integrins, New York-London, Springer, 1997.
Erickson, Couchman 2000: Erickson, Anna C. - Couchman, John R., Still more complexity in mammalian basement membranes, "Journal of histochemistry and cytochemistry", 48, 2000, pp. 1291-1306.
Hohenester, Engel 2002: Hohenester, Erhard - Engel, Jürgen, Domain structure and organisation in extracellular matrix proteins, "Matrix biology", 21, 2002, pp. 115-128.
Kielty 2002: Kielty, Cay M. - Sherratt, Michael J. - Shuttleworth, Adrian, Elastic fibres, "Journal of cell science", 115, 2002, pp. 2817-2828.
Kyriakides, Bornstein 2003: Kyriakides, Themis R. - Bornstein, Paul, Matricellular proteins as modulators of wound healing and the foreign body response, "Thrombosis and haemostasis", 90, 2003, pp. 986-992.
Lodish 1995: Molecular cell biology, 3. ed., edited by Harvey Lodish e altri, New York, Scientific American Books, 1995.
Mainiero 1997: Mainiero, Fabrizio e altri, The coupling of alpha6beta4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation, "EMBO journal", 16, 1997, pp. 2365-2375.
Mainiero 1998: Mainiero, Fabrizio - Santoni, Angela, Fibronectin, in: Encyclopedia of immunology, 2. ed., edited by Peter J. Delves and Ivan M. Roitt, San Diego-London, Academic Press, 1998.
Mainiero 2000: Mainiero, Fabrizio e altri, Integrine e cellule natural killer, "EOS-Journal of immunology and immunopharmacology", 20, 2000, pp. 63-68.
McGowan, Marinkovich 2000: McGowan, Kelly A. - Marinkovich, M. Peter, Laminins and human disease, "Microscopy research and technique", 51, 2000, pp. 262-279.
Myllyharju, Kivirikko 2004: Myllyharju, Johanna - Kivirikko, Kari, Collagens, modifying enzymes and their mutations in humans, flies and worms, "Trends in genetics", 20, 2004, pp. 33-43.
Ramirez 2004: Ramirez, Francesco e altri, Fibrillin microfibrils: multipurpose extracellular networks in organismal physiology, "Physiological genomics", 19, 2004, pp. 151-154.
Ricard-Blum, Ruggiero 2005: Ricard-Blum, Sylvie - Ruggiero, Florence, The collagen superfamily: from the extracellular matrix to the cell membrane, "Pathologie-biologie", 53, 2005, pp. 430-432.
Sanders 1998: Sanders, Robert J. - Mainiero, Fabrizio - Giancotti Filippo G., The role of integrins in tumorigenesis and metastasis, "Cancer investigation", 16, 1998, pp. 329-344.
Seiffert 1997: Seiffert, Dietmar, Constitutive and regulated expression of vitronectin, "Histology and histopathology", 12, 1997, pp. 787-797.
Tunggal 2000: Tunggal, Patrick e altri, Laminins: structure and genetic regulation, "Microscopy research and technique", 51, 2000, pp. 214-227.
Wagener 2005: Wagener, Raimund e altri, The matrilins - adaptor proteins in the extracellular matrix, "FEBS letters", 579, 2005, pp. 3323-3329.
Siti internet
http://www.retemalattierare.it/
http://www.orpha.net/consor/cgi-bin/home.php?Lng=IT
http://www.telethon.it/informagene/malattie.asp
http://www.mucopolisaccaridosi.it/
http://medical-dictionary.thefreedictionary.com
http://www.geocities.com/CapeCanaveral/9629/index.html