
Proteine. Struttura delle proteine
Proteine. Struttura delle proteine
Le proteine, sintetizzate come catene polipeptidiche che si estendono in modo spazialmente non strutturato, devono raggiungere una conformazione tridimensionale stabile per poter svolgere le loro funzioni biologiche. Da decenni è noto che tutte le informazioni necessarie a un polipeptide denaturato per il raggiungimento di una conformazione stabile e funzionale sono contenute all'interno della sua stessa sequenza amminoacidica. In teoria, quindi, la conformazione biologicamente attiva di una proteina dovrebbe essere desumibile una volta che sia nota la sua struttura primaria. In pratica, però, sono ancora ignote le regole che ci permetterebbero di predire la conformazione spaziale di una proteina a partire dalla conoscenza della sequenza lineare dei suoi amminoacidi. Sebbene, in vitro, sia stato dimostrato che numerose proteine sono in grado di assumere spontaneamente una struttura terziaria, in vivo, tuttavia, il processo che porta all'assunzione di una corretta conformazione sterica coinvolge un complesso intreccio di interazioni fra tre diverse molecole.
L'insieme delle modifiche conformazionali che in una proteina caratterizzano i passaggi da una struttura lineare a una struttura tridimensionale è denominato 'folding' (ripiegamento). La comprensione dei meccanismi utilizzati dai polipeptidi per raggiungere una struttura tridimensionale che li renda in grado di svolgere correttamente le loro funzioni biologiche è stata in questi ultimi anni notevolmente favorita da numerosi studi sulla chimica fisica e sulla biologia cellulare di questo processo. Sebbene la conoscenza di tale argomento sia lontana dall'essere completa, è, tuttavia, opinione generale che il folding proceda attraverso molteplici passaggi distinti. Durante questo processo biologico altamente complesso si devono infatti formare e rompere numerose interazioni all'interno della catena polipeptidica in fase di ripiegamento e, in vivo, tra essa e gli enzimi e le proteine che facilitano questo processo. Per la funzione che svolgono, queste ultime sono dette 'chaperon molecolari'.
Dopo la spiegazione del ruolo del codice genetico, lo studio del folding delle proteine favorirà notevolmente la comprensione del modo in cui la natura riesce a trasformare l'informazione genetica monodimensionale in strutture biologiche specifiche. Un eventuale successo nella scoperta di un codice di folding si tradurrebbe in un salto qualitativo d'informazione per i giganteschi progetti sui genomi che producono continuamente sequenze di DNA a un ritmo via via crescente.
Pertanto la comprensione dei meccanismi e delle vie che conducono al folding potrebbe rivelarsi uno strumento di incalcolabile valore nella traduzione di sequenze amminoacidiche in strutture tridimensionali.
Lo stato di prefolding
Idealmente lo stato privo di folding è il cosiddetto random coil (catena attorcigliata a caso), in cui le conformazioni possibili, anche per una proteina di piccole dimensioni, sono moltissime. In questa fase il polipeptide si dovrebbe trovare in uno stato in cui la sua catena è molto estesa nello spazio e le interazioni non covalenti, che normalmente stabilizzano lo stato nativo, sono inesistenti. È lecito supporre che le proteine, sintetizzate come polipeptidi lineari sui ribosomi, in vivo comincino il folding durante la loro stessa sintesi. Tuttavia evidenze sperimentali suggeriscono che le proteine destinate a essere veicolate a particolari compartimenti cellulari, quali cloroplasti, mitocondri e reticolo endoplasmatico, passino attraverso le membrane intracellulari in una conformazione estesa o con folding lasso. Si pensa che a mantenere le proteine neosintetizzate in tale stato di conformazione lassa, necessario per la loro veicolazione verso i vari organelli cellulari, siano le chaperon molecolari.
Le strutture globulari semisolide
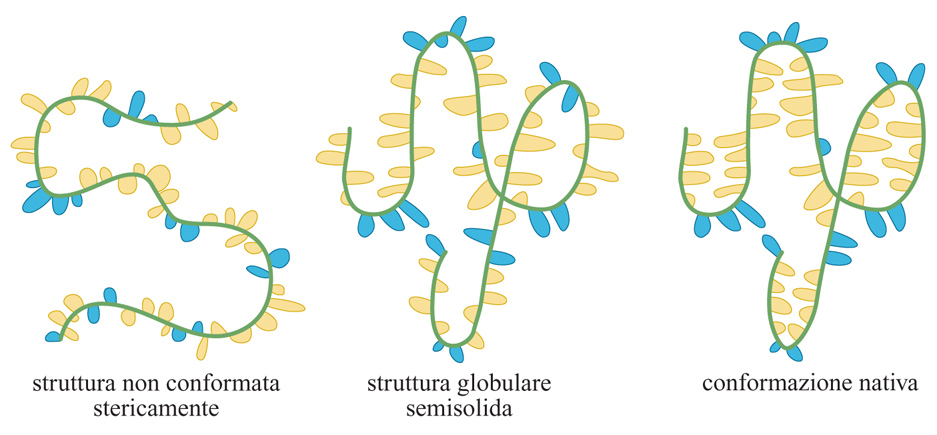
È stato osservato che molte proteine, in certe condizioni, si trovano in una conformazione stabile che non è di folding compiuto, ma neanche di completa disorganizzazione sterica (fig. 2). Il verificarsi di conformazioni globulari semisolide (molten globule) è ben documentato. Le proprietà caratteristiche di tali strutture sono: la maggior compattezza rispetto allo stato di random coil e la dimensione lievemente maggiore rispetto alla proteina nativa; un contenuto in strutture secondarie simile a quello della proteina con folding completo; la presenza di superfici idrofobiche esposte all'esterno, che le rendono suscettibili di aggregazione reciproca; un'entalpia pressoché identica a quella che esse stesse hanno in uno stato privo di folding; un'interconversione, dalla condizione di strutture globulari semisolide allo stato di completa disorganizzazione sterica, rapida e non cooperativa, e invece passaggio allo stato di folding completo lento e cooperativo. Queste osservazioni suggeriscono che la struttura globulare semisolida sia una molecola collassata, dotata di strutture secondarie simili a quelle della corrispondente proteina nativa, ma priva di strutture terziarie stabili. Il significato biologico delle strutture globulari semisolide è oggetto di discussione.
Lo stato nativo
Lo stato nativo di molte proteine diverse è stato esaminato con la cristallografia a raggi X e con l'analisi spettroscopica NMR. Questi studi hanno chiarito alcuni aspetti generali delle proteine globulari il cui folding sia avvenuto compiutamente e che si trovano quindi nello stato nativo. La caratteristica che accomuna tutte queste proteine globulari è la non polarità delle catene laterali che formano la parte interna della struttura e la generale prevalenza di catene laterali idrofiliche esposte alla superficie. Per la maggior parte delle proteine non si conosce che un unico stato di folding completo. Le interazioni che stabilizzano lo stato nativo sono intrinsecamente deboli, ma la presenza contemporanea di un gran numero di tali interazioni e la loro cooperatività hanno come risultato la produzione di strutture stabili.
4. Le vie di folding in vitro e in vivo
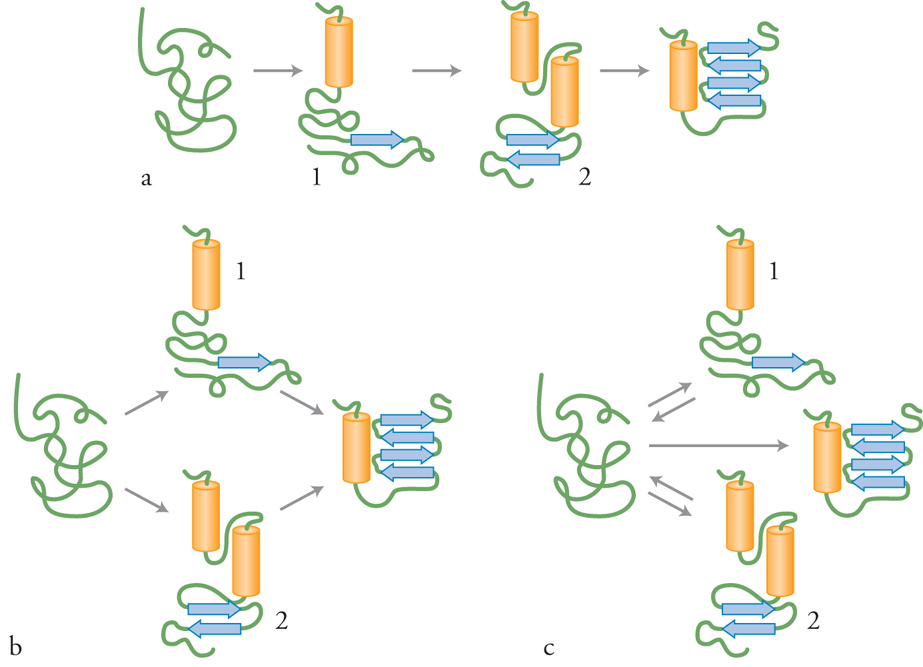
Si ritiene che il refolding in vitro avvenga attraverso diverse vie che coinvolgono uno o più stati intermedi di folding relativamente stabili. Questi ultimi sembrano trovarsi in un equilibrio rapido con lo stato di denaturazione completa (nell'ordine di millisecondi), mentre sono convertiti allo stato nativo solo molto lentamente. Si ritiene che la conversione degli intermedi di folding nello stato nativo sia un processo cooperativo in cui il verificarsi di un'interazione rende stabile la successiva, che a sua volta stabilizzerà la prima interazione. La semplice via lineare di folding che procede attraverso la formazione di una serie di intermedi, uno successivo all'altro (fig. 3), potrebbe non essere applicabile alla maggior parte dei polipeptidi. Esistono varie prove dell'esistenza di vie di folding multiple e indipendenti. Secondo questa ipotesi, il folding di una proteina denaturata potrebbe avvenire attraverso vie distinte e parallele che comportano l'insorgenza di forme intermedie diverse ma che portano a un unico stato nativo ben definito (fig. 3B). È sorto il dubbio che gli intermedi osservati non rappresentino stati reali del folding, ma che siano invece prodotti di reazioni collaterali non produttive che non conducono allo stato nativo (fig. 3C). Questo dubbio è scaturito dalla scoperta che il folding di alcune piccole proteine avviene in modo estremamente veloce e secondo una modalità 'tutto o nulla' in cui non sono identificabili intermedi di folding.
Gli esperimenti in vitro hanno sicuramente un valore significativo nel definire i tipi di interazioni intramolecolari che guidano il folding delle proteine, ma non riflettono accuratamente i processi di folding delle proteine nascenti all'interno della cellula. Le proteine non completamente strutturate, o in stati intermedi di folding, tendono a esporre superfici idrofobiche all'ambiente acquoso circostante e sono quindi particolarmente inclini all'aggregazione. In vivo la temperatura fisiologica e l'alta concentrazione sia delle proteine totali sia dei polipeptidi non strutturati favoriranno di gran lunga le interazioni improduttive di aggregazione rispetto alla via di folding corretta. Un'ulteriore differenza tra il folding delle proteine in vitro e in vivo è il livello di complessità di molte proteine all'interno delle cellule. Proteine di membrana altamente idrofobiche, proteine che si assemblano in complessi enzimatici o in microfilamenti e proteine che sono modificate durante o dopo il processo di traduzione mediante legami con lipidi o carboidrati difficilmente seguiranno le vie di folding valide per piccole proteine globulari. Un'ulteriore considerazione è che, nella cellula, le proteine sono sintetizzate per gradi sui ribosomi e, nel caso di proteine dirette all'interno degli organelli, sono traslocate attraverso le membrane in forma estesa; esse dovrebbero quindi, secondo la teoria del folding cooperativo, permanere in forma denaturata almeno fino al compimento della sintesi del primo dominio strutturale. Inoltre si è visto che alcune tappe lente nel folding delle proteine sono catalizzate da enzimi specifici. Negli ultimi anni è divenuto sempre più chiaro il modo in cui le cellule affrontano questi problemi di folding.
5. Enzimi coinvolti nel foldingdelle proteine
Due passaggi cruciali per la cinetica del processo di folding in vitro, consistenti nell'isomerizzazione di legami covalenti, possono essere catalizzati da enzimi cellulari purificati. L'enzima proteindisolfuroisomerasi (PDI) catalizza lo scambio tiolo-disolfuro e promuove la formazione, l'isomerizzazione o la riduzione di ponti disolfuro all'interno delle proteine. Gli enzimi peptidil prolil cis-trans isomerasi (PPIasi) catalizzano l'altrimenti lenta isomerizzazione dei legami peptidici che precedono i residui amminoacidici di prolina. Ambedue questi enzimi non determinano la via di folding dei polipeptidi, ma piuttosto accelerano il folding di polipeptidi che formano ponti disolfuro o sono ricchi in prolina.
La proteindisolfuroisomerasi
La formazione non catalizzata di legami disolfuro è una tappa lenta nel processo di folding di molte proteine e PDI accelera questa reazione. Poiché solo le proteine della via secretiva contengono legami disolfuro, PDI è localizzato essenzialmente in compartimenti cellulari che sono parte integrante di tale via. Grazie alla presenza di questo enzima, in vivo la formazione di legami disolfuro corretti può essere molto rapida. In alcuni casi, come per le catene peptidiche delle immunoglobuline, è addirittura un evento cotraduzionale, cioè avviene prima che la sintesi della catena polipeptidica sul ribosoma sia terminata: appena un intero dominio proteico viene traslocato nel lume del reticolo endoplasmatico, si forma contemporaneamente il singolo legame disolfuro interno a quel dominio. Il ruolo di PDI appare ora abbastanza chiaro: esso non determina la via di folding ma piuttosto catalizza passaggi cineticamente lenti. La direzione in cui avviene il folding e il prodotto finale sono invece determinati dalla proteina stessa, cioè dal corredo nativo di legami disolfuro e da appropriate condizioni di ossidoriduzione.
Le peptidilprolilcis-transisomerasi
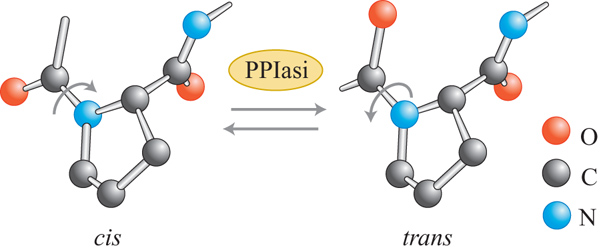
I legami peptidici possono esistere sia in forma cis sia in forma trans. Generalmente, dal punto di vista energetico, la forma trans è 10.000 volte più stabile della forma cis. La situazione è diversa per i legami peptidici tra un amminoacido qualunque e la prolina: in questo caso la forma trans è pur sempre favorita, ma solo di 4 volte. L'isomerizzazione cis-trans spontanea dei legami peptidici prolinici è lenta. L'isomerizzazione dei legami peptidici amminoacido-prolina non corretti è, perciò, uno di quei passaggi lenti cruciali nel determinare la cinetica di folding di una proteina nella cellula. La reazione può essere catalizzata da enzimi con attività tipo PPIasica. Le PPIasi sono molto abbondanti, distribuite in modo quasi ubiquitario e sono presenti in tutti i compartimenti cellulari. Studi in vitro sulle PPIasi hanno mostrato che tali enzimi accelerano il refolding di un ampio spettro di proteine, ma con efficienza variabile. I legami prolinici situati all'interno degli intermedi di folding non sono tuttavia accessibili alle PPIasi. La reazione di folding delle proteine che viene catalizzata da questi enzimi è una rotazione di 180° attorno all'asse C-N del legame peptidico che precede la prolina, che non coinvolge né scissione né formazione di legami covalenti. Perciò le PPIasi possono essere classificate come 'conformasi' (cioè enzimi in grado di modificare la conformazione molecolare) dotate di un'altissima efficienza (fig. 4).
6. Chaperon molecolari
Alcune famiglie di proteine, strutturalmente non correlate ma universalmente conservate, aiutano le proteine neosintetizzate ad assumere la loro conformazione nativa. Queste proteine, ora collettivamente denominate 'chaperon' molecolari, si legano ai polipeptidi quando si trovano nello stato totalmente o parzialmente denaturato, impedendone così l'aggregazione. Le chaperon molecolari sono piuttosto abbondanti e l'espressione di molte di esse aumenta notevolmente in diverse condizioni di stress della cellula. Per ragioni storiche molte di queste chaperon molecolari sono classificate come 'proteine dello shock da calore' (Hsp, Heat shock proteins) o proteine da stress. Nonostante il legame e il rilascio da alcune Hsp siano regolati dal legame e dall'idrolisi dell'ATP, le chaperon molecolari non possono essere considerate come catalizzatori del folding delle proteine. Più probabilmente esse impediscono l'aggregazione e le interazioni improduttive, creando un ambiente regolato e protetto affinché il folding possa avvenire in maniera corretta e con maggiore efficienza.
La maggioranza delle chaperon identificate finora appartiene a famiglie proteiche molto conservate. Nonostante siano state identificate numerose classi di chaperon, noi ci occuperemo solo delle famiglie di chaperon Hsp60 (GroEL) e Hsp70 (DnaK), che sono quelle maggiormente studiate (i numeri indicano la massa molecolare approssimativa, in migliaia di dalton, dei membri di ciascuna famiglia). Negli ultimi anni è stato chiarito che queste principali chaperon intracellulari non agiscono da sole. Per assicurare la loro corretta ed efficiente funzione, si sono evolute le cosiddette co-chaperon. In alcuni casi queste co-chaperon partecipano direttamente all'azione delle chaperon, come DnaJ (Hsp40) di Escherichia coli, che agisce sinergicamente con DnaK. In altri casi, hanno un ruolo più indiretto assicurando la corretta funzione e il riciclaggio delle proteine chaperon.
Prima di analizzare le proprietà dei differenti complessi di chaperon, ci sembra necessario esporre le nostre idee sul meccanismo molecolare attraverso il quale le chaperon svolgono la loro funzione. In primo luogo bisogna distinguere tra 'facilitazione' di un processo e 'catalisi'. Durante il folding, i polipeptidi procedono attraverso stati generalmente separati solo da barriere energetiche molto basse. Proteine piccole, con un solo dominio, spesso assumono il folding corretto molto velocemente senza che nessun intermedio cinetico possa essere fissato. Proteine più grandi iniziano la tappa finale del folding a partire da un intermedio che è separato dallo stato nativo da una barriera energetica molto alta. Il passaggio finale è quello che limita la velocità della reazione ed è fortemente cooperativo. La velocità con cui appare il prodotto nativo dipenderà dalla concentrazione di questo intermedio e dalla costante di velocità. La costante di velocità è correlata alla barriera energetica che c'è tra l'intermedio e lo stato finale (energia di attivazione). La catalisi enzimatica, dovuta agli enzimi descritti prima, aumenta la costante di velocità abbassando questa barriera energetica. Le chaperon molecolari, invece, facilitano il folding senza aumentarne la velocità. Esse aumentano la 'quantità' di proteina correttamente conformata. Infatti, le chaperon, impedendo l'aggregazione, aumentano la resa della reazione di folding.
Abbondanza, velocità di legame e specificità
L'aggregazione, poiché comporta contatti tra due o più molecole, è ovviamente un processo almeno di secondo ordine e, quindi, la velocità di aggregazione è molto sensibile alla concentrazione degli intermedi di folding. Il folding, invece, è un processo di primo ordine e, quindi, molto meno influenzato dalla concentrazione. Di conseguenza, un incremento della concentrazione degli intermedi di folding liberi porterà a un aumento dell'aggregazione, mentre una diminuzione della loro concentrazione favorirà il folding. Una chaperon potrebbe agire diminuendo la concentrazione degli intermedi liberi. Se il legame alle chaperon deve efficientemente competere con l'aggregazione, la velocità di legame dovrebbe avvicinarsi al limite di incontro tra chaperon e polipeptide, ed è stato calcolato che questo accade. La chaperon potrebbe quindi efficacemente contrastare l'aggregazione. Caratteristica comune delle chaperon è la capacità di riconoscere polipeptidi solo allo stato non nativo. Questo riconoscimento deve portare a un forte e rapido legame del ligando e al sequestro delle sue superfici idrofobiche così da prevenirne l'aggregazione.
7. La famiglia delle Hsp70
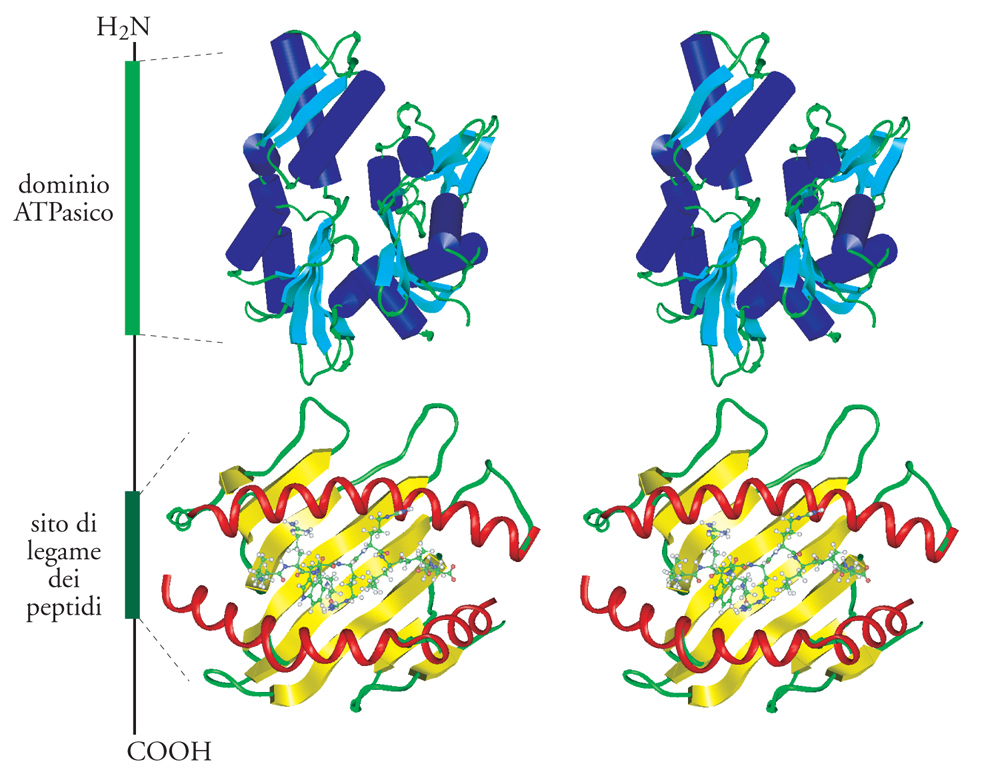
Membri della famiglia delle Hsp70 si trovano in tutte le cellule procariotiche ed eucariotiche, nel citoplasma, nel nucleo, nel reticolo endoplasmatico, nei mitocondri e nei cloroplasti. Alcune proteine di questa grande famiglia sono essenziali per la crescita cellulare, altre sono copiosamente espresse in seguito a shock da calore, altre ancora, chiamate Hsc (Heat shock cognate) sono espresse, invece, in modo costitutivo. Tutti i membri della famiglia delle Hsp70 recano all'estremità amminoterminale un dominio ATPasico altamente conservato seguito da una porzione carbossiterminale, meno conservata, che contiene il sito di legame dei peptidi (fig. 5). Alcuni membri della famiglia possiedono, inoltre, una sequenza segnale amminoterminale o una sequenza carbossiterminale di ritenzione nel reticolo endoplasmatico, a seconda della destinazione finale della proteina Hsp70 in questione. La proteina Hsc70 bovina è dotata di quattro domini strutturali che formano due lobi con in mezzo una fenditura. La tasca di legame per il nucleotide ATP e uno ione magnesio è situata alla base della fenditura. I residui amminoacidici che interagiscono con il complesso nucleotide-Mg2+ nel sito attivo sono rigorosamente conservati attraverso tutta la famiglia delle Hsp e, pertanto, si può suggerire un loro ruolo nel controllo dell'attività ATPasica delle proteine. Un frammento di 18 kDa, situato immediatamente dopo il dominio ATPasico, è sufficiente per il legame ad alta affinità del substrato.
Studi in risonanza magnetica nucleare di peptidi legati alle Hsp70 suggeriscono che il prerequisito fondamentale per l'interazione non sia tanto l'idrofobicità del substrato quanto l'assenza di una struttura secondaria. Il modello per l'azione delle Hsp70 è costituito dalla proteina DnaK di E. coli, che agisce di concerto con altre due Hsp, DnaJ e GrpE. Sia DnaJ che DnaK sono in grado di legare polipeptidi, ma la loro affinità per i substrati è diversa. Sembra che, mentre DnaK preferisce interagire con proteine non strutturate stericamente, DnaJ tenda a legare substrati proteici che contengono strutture secondarie e terziarie. Apparentemente queste differenze in specificità di substrato permettono a proteine tipo DnaJ di facilitare l'interazione di proteine nascenti con membri della famiglia Hsp70, che altrimenti non verrebbero legate. La DnaJ di E. coli riconosce una caratteristica strutturale tipica degli intermedi di folding, li stabilizza in una conformazione in cui sono esposti i siti di legame per le Hsp70 e facilita in questo modo il legame del substrato con DnaK. DnaJ agisce anche sulla stessa DnaK stimolando il passaggio chimico limitante nel ciclo di reazione ATPasico di DnaK. A differenza di DnaJ, GrpE promuove lo scambio nucleotidico agendo come un fattore di rilascio dei nucleotidi. GrpE interagisce con un'ansa strutturalmente conservata vicina al sito di legame per l'ATP di DnaK e presumibilmente tramite questa interazione influenza le condizioni di legame dei nucleotidi della chaperon.
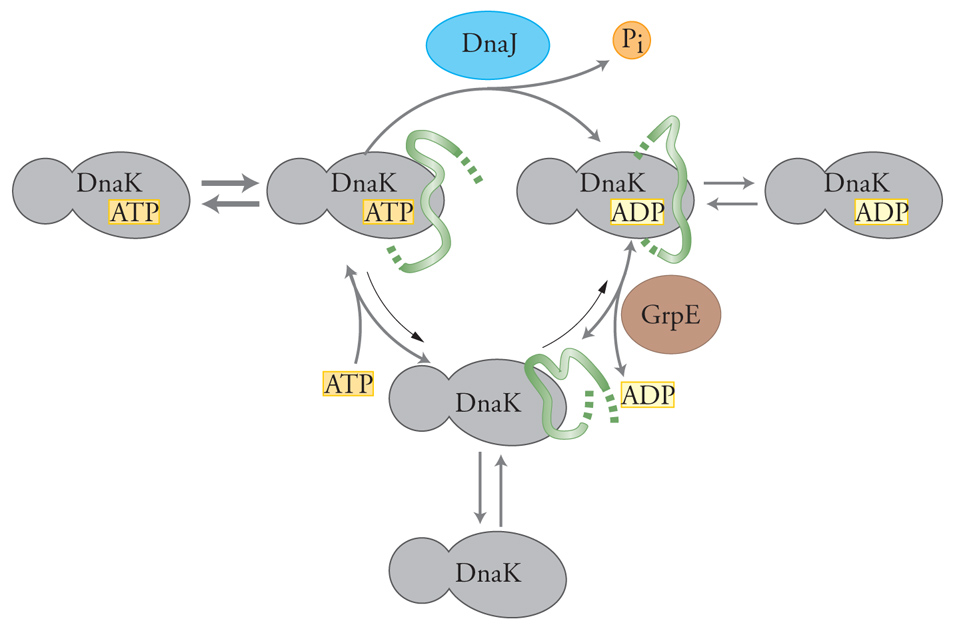
Studi recenti hanno chiarito alcuni aspetti importanti del ciclo ATPasico di DnaK. Da questi risultati è emerso un modello generale per il ciclo di reazioni delle proteine Hsp70 (fig. 6). Il legame dell'ATP alle Hsp70 accelera sia la velocità di legame sia il rilascio del substrato peptidico. Le Hsp70, nella loro conformazione con ATP legato, possono essere quindi definite come la forma a 'pronto legame-pronto rilascio'. Le Hsp70, nella loro conformazione con ADP legato, rilasciano i polipeptidi solo lentamente e, quindi, tale conformazione è stata all'inizio definita con l'espressione di 'stato di alta affinità'; ma in realtà è molto lenta anche la formazione del legame con i peptidi. La conformazione con ADP legato può essere quindi definita come la forma a 'lento legame-lento rilascio'. La presenza di GrpE induce il rilascio dell'ADP generando una conformazione priva di nucleotidi. Il legame dell'ATP completa il ciclo e porta al rilascio della catena polipeptidica. La conformazione di Hsp70 risultante, cioè quella legata all'ATP, sarà di nuovo pronta per un altro ciclo di legame e il rilascio di un nuovo polipeptide.
8. Le chaperon Hsp70 nella biogenesi delle proteine mitocondriali
Un elegante esempio delle differenti funzioni che le Hsp70 svolgono durante la sintesi e la maturazione delle proteine è dato dalla biogenesi delle proteine mitocondriali nel loro passaggio dal sito di sintesi nel citosol alla localizzazione funzionale nei mitocondri. Le proteine mitocondriali sono sintetizzate su ribosomi citosolici come precursori, contenenti una sequenza segnale all'estremità amminoterminale. Esse vengono quindi trasportate ai mitocondri, traslocate in una conformazione estesa attraverso le due membrane mitocondriali, per assumere infine la conformazione nativa nella matrice mitocondriale. Le chaperon Hsp70 influenzano tutti gli stadi di questo processo e sono direttamente coinvolte nella traslocazione del polipeptide attraverso le membrane mitocondriali.
Il ruolo delle Hsp70 durante la sintesi delle proteine
Il genoma del lievito Saccharomyces cerevisiae codifica dieci diverse proteine Hsp70. Ci sono tre sottofamiglie citosoliche: SSA con quattro membri, SSB con due, SSE con due, per un totale di otto geni. I rimanenti due geni, SSC1 e KAR2, codificano rispettivamente una proteina Hsp70 mitocondriale e una Hsp70 localizzata nel reticolo endoplasmatico. Le sottofamiglie citosoliche hanno ruoli diversi nella cellula. I membri codificati da SSA sono implicati nel trasporto proteico e hanno un ruolo nella regolazione della risposta al calore e nella regolazione della degradazione proteica. Si pensa, invece, che i membri codificati da SSB siano coinvolti nella sintesi proteica. Le proteine Ssb si associano ai ribosomi attivamente coinvolti nella sintesi proteica dai quali vengono rilasciate dopo trattamento con puromicina, un antibiotico che causa il rilascio delle catene polipeptidiche nascenti. Si potrebbe immaginare che una proteina Ssb si leghi a una catena nascente appena questa emerge dalla subunità ribosomale 60S, impedendo le interazioni intramolecolari all'interno della catena stessa o tra la catena nascente e la superficie del ribosoma e facilitando, quindi, la fuoriuscita dal ribosoma del resto della catena nel corso della sua sintesi.
Il ruolo delle Hsp70 nel trasporto delle proteine all'organello bersaglio
Le proteine, per attraversare le membrane biologiche, devono avere una conformazione lassa. Dati sperimentali scaturiti dallo studio della traslocazione delle proteine nei mitocondri hanno evidenziato la necessità che, durante questi processi, i precursori delle proteine, per essere competenti per la traslocazione, siano in uno stato particolare e non nativo. Le chaperon molecolari della famiglia Hsp70 aiutano a mantenere nello stato competente per la traslocazione le proteine destinate ai mitocondri, al reticolo endoplasmatico, ai cloroplasti e al nucleo. Nel lievito questa funzione è espletata dalle proteine Ssa. Essenziale in questo stadio è che le chaperon Hsp70 siano legate al precursore della proteina da traslocare, in modo da prevenire il folding nello stato nativo. In questo caso è evidente il significato dell'assunzione di una conformazione a rilascio lento da parte delle Hsp citoplasmatiche. Nel trasporto ai mitocondri le preproteine sono inizialmente trasferite a recettori specifici posti sulla superficie della membrana mitocondriale esterna, i quali, a loro volta, presentano la proteina al canale attraverso cui avverrà la traslocazione vera e propria.
Il ruolo delle Hsp70 nella traslocazione delle proteine
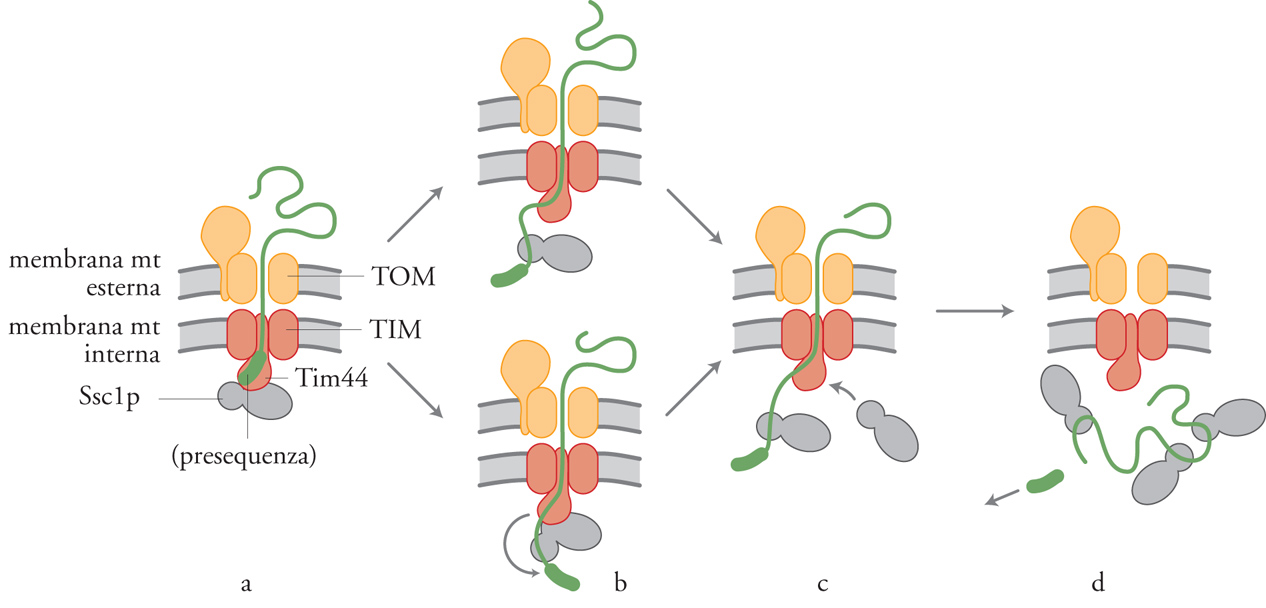
La traslocazione dei precursori proteici attraverso le due membrane mitocondriali richiede sia ATP sia un potenziale elettrico a cavallo della membrana mitocondriale interna. Si pensa che il potenziale di membrana fornisca la forza trainante per la traslocazione della sequenza segnale della proteina mitocondriale, mentre la richiesta di ATP è attribuita prevalentemente al funzionamento della Hsp70 mitocondriale (Scc1p nel lievito). Esperimenti su diversi mutanti ssc1 in cui veniva tolto ATP dalla matrice mitocondriale hanno permesso di stabilire che per completare la reazione di traslocazione è necessario che Ssc1 si leghi ai precursori delle proteine mitocondriali (preproteine) che stanno entrando nel mitocondrio. Al momento sono diffusi due modelli che descrivono l'azione di Scc1p (fig. 7). Nel primo modello, chiamato Brownian ratchet (o 'modello del nottolino browniano'), Ssc1p ha un ruolo più passivo durante il processo di traslocazione. Per moto browniano la proteina in conformazione lassa scivola avanti e indietro attraverso il canale ed è intrappolata da Ssc1p dal lato della matrice. Secondo questo modello, Ssc1p avrebbe quindi un'attività di bloccaggio. Il modello del nottolino browniano non può, però, spiegare facilmente la scoperta che alcuni precursori vengono attivamente estesi da Ssc1p durante la traslocazione nei mitocondri. È stato perciò proposto un secondo modello nel quale Ssc1p avrebbe un ruolo più attivo. Il precursore che sta entrando è legato da Ssc1p dal lato della matrice ed è poi trascinato all'interno del mitocondrio mediante un cambio conformazionale di Ssc1p che rimane legata sia al precursore sia alla membrana. Questo cambio conformazionale genererebbe una forza che potrebbe permettere lo srotolamento ('unfolding') del precursore e la sua traslocazione all'interno del mitocondrio.
Il ruolo di Hsp70 nel folding delle proteine traslocate
In seguito alla traslocazione nel mitocondrio e alla rimozione della presequenza, le proteine legate a Ssc1p devono assumere la conformazione nativa. Quindi, mentre le molecole Hsp70 sul lato citoplasmatico devono impedire il folding dei precursori proteici, sul lato della matrice proteine molto simili alle Hsp70 devono facilitare questo processo. Tale funzione viene svolta grazie alla cooperazione con un membro dell'altra importante famiglia delle chaperon, quella delle Hsp60, note anche con il nome di chaperonine.
9. Le chaperonine
Le chaperonine sono una famiglia di molecole chaperon formate da subunità di circa 60 kDa, che si assemblano in oligomeri di quattordici subunità dalla caratteristica forma a doppio anello. Le chaperonine sono estremamente conservate e si trovano negli eubatteri (GroEL), nei mitocondri (Hsp60) e nei cloroplasti (RuBisCO binding protein, proteina legante la RuBisCO). Le chaperonine costituiscono così un sottogruppo delle proteine chaperon molecolari. La funzione di GroEL è correlata a quella della chaperonina GroES, la cui struttura cristallina è stata chiarita solo di recente. GroES è una proteina essenziale, composta di un singolo anello formato da sette subunità di 10 kDa. Questa proteina forma un complesso asimmetrico con GroEL, in un rapporto stechiometrico di 1:1, legandosi a un'estremità della struttura cilindrica di GroEL. Substrati polipeptidici non conformati stericamente si legano a GroEL nello stesso dominio riconosciuto da GroES, mentre il sito di legame dell'ATP è situato sulla superficie più interna del dominio equatoriale, prossimo al centro del cilindro. Un cambiamento conformazionale potrebbe quindi accoppiare la presenza del nucleotide appropriato con il processo, assistito da GroES, di legame e rilascio dei polipeptidi.
GroEL può formare un complesso molto forte con varie proteine che si trovano in uno stato strutturale non conformato, mentre ha poca affinità per le proteine allo stato nativo. GroEL e GroES, a concentrazioni fisiologiche di ATP e ADP, formano un complesso stabile ma dinamico. Il complesso asimmetrico GroEL-GroES, quando è legato all'ADP, agisce probabilmente come accettore di polipeptidi poiché si tratta di una specie conformazionale dotata di un'esistenza relativamente lunga e di un'alta affinità per i polipeptidi. Un'estremità del cilindro GroEL è 'incappucciata' da GroES lì legata (il lato cis) e ciò dovrebbe impedire l'accesso di una proteina alla regione di legame del peptide, situata nella cavità centrale posta a quella estremità del cilindro. I polipeptidi potrebbero, quindi, legarsi inizialmente all'anello di GroEL non occupato da GroES (il lato trans). Durante il ciclo di folding, il rilascio e il successivo legame di GroES permetterebbero la formazione di un complesso cis nel quale sia GroES sia il polipeptide sono legati allo stesso anello di GroEL. GroES continuerebbe a svolgere la sua funzione di incappucciato, ma questa volta, invece di escludere il polipeptide dalla cavità centrale di GroEL, lo manterrebbe sequestrato al suo interno. La transizione da un complesso trans a uno cis con una sola molecola di GroES legata ('modello a pallottola') potrebbe comprendere una fase in cui il complesso è privo di GroES o, in alternativa, il complesso ha un cappuccio GroES ad ambedue i lati del cilindro, generando la struttura intermedia rugby.
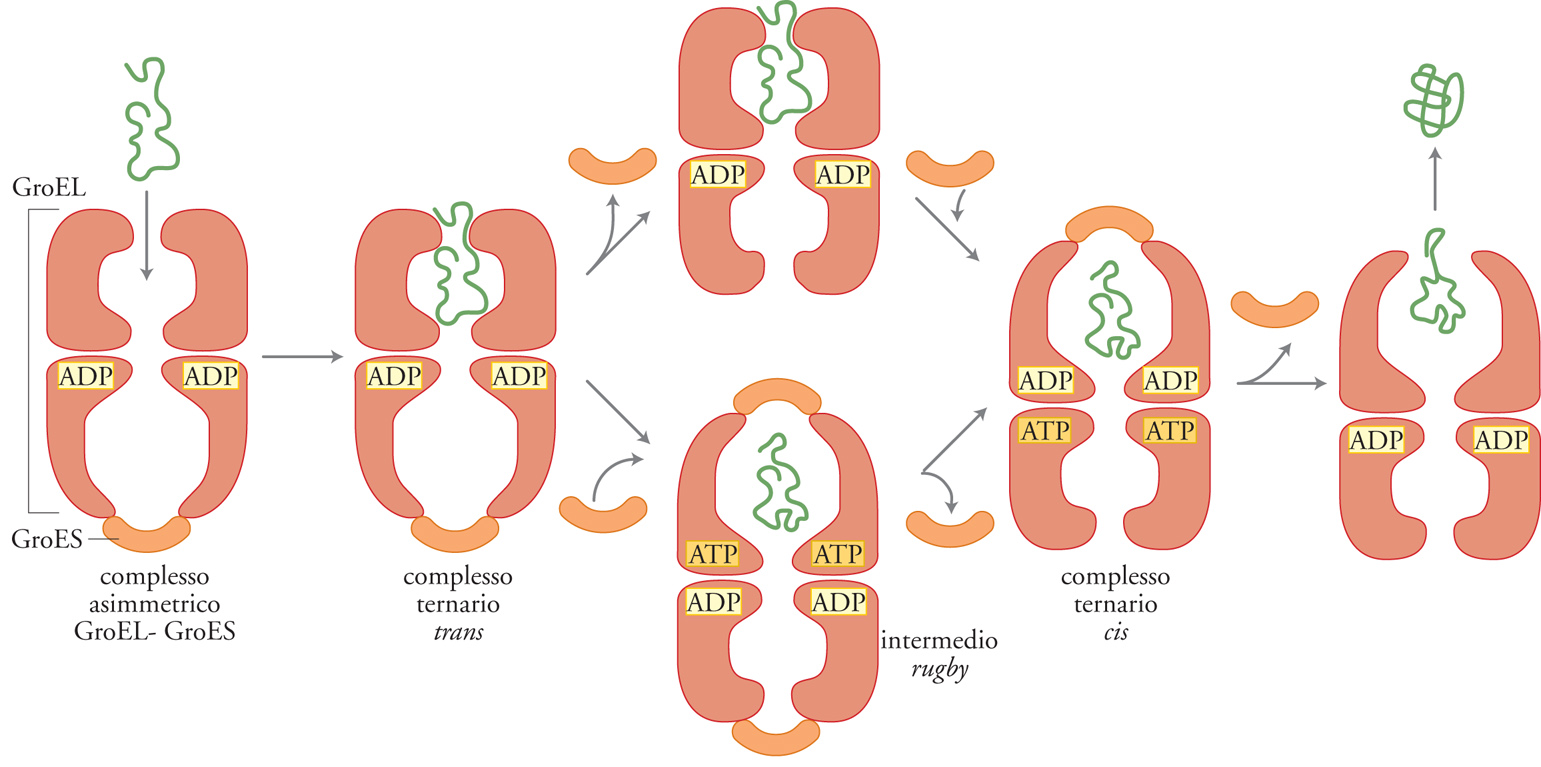
Il legame e l'idrolisi di ATP nell'anello di GroEL, opposto a quello occupato da GroES e dal polipeptide, indurrebbero il rilascio di GroES legato al lato cis e permetterebbero il liberarsi del polipeptide nel citosol. Dal modello appena descritto del meccanismo d'azione del complesso GroEL-GroES consegue che ci deve essere un limite fisico alle dimensioni dei polipeptidi assistiti dalle chaperonine nel folding. In assenza di co-chaperonine, peptidi di dimensioni fino a circa 35 kDa possono entrare dentro un singolo anello di GroEL. Il legame della co-chaperonina GroES induce, tuttavia, in GroEL un cambiamento conformazionale di vaste proporzioni: il raddoppio quasi del volume della cavità centrale dell'anello (fig. 8). Sebbene questo modello abbia chiarito che le chaperonine agiscono secondo una modalità regolata, legando e rilasciando polipeptidi non conformati stericamente, non è ovvio come questo ciclo di legame e rilascio porti a un aumento dell'efficienza del folding. Attualmente sono argomento di intensa discussione due punti di vista contrastanti. Alcuni ritengono che il polipeptide sia estruso dalla chaperonina in una conformazione ancora non strutturata stericamente e che il folding finale avvenga all'esterno della chaperonina, nel citosol. Una teoria alternativa sostiene che il polipeptide vada incontro al folding all'interno del complesso GroEL-GroES e che sia estruso dalla chaperonina solo quando non si possano più verificare eventi di interazione tra il complesso e il polipeptide.
Secondo il primo modello le chaperonine potrebbero agire attuando un sequestro degli intermedi di folding, riducendo quindi la concentrazione di quei polipeptidi tendenti all'aggregazione: la competizione con l'aggregazione porterebbe, in questo caso, a un aumento netto del folding. Alcune osservazioni sperimentali suggeriscono che il meccanismo non sia esclusivamente passivo ma che, in certe circostanze, le chaperonine possano abbassare in modo attivo la barriera di energia del folding, fungendo quindi da veri e propri enzimi. La prima evidenza è che il folding della malicodeidrogenasi è assistito da GroEL a rapporti stechiometrici molto bassi, pari a una molecola di GroEL per ogni dieci molecole di deidrogenasi. In queste condizioni il 90% del substrato deve essere libero in soluzione, così che l'effetto del sequestro di polipeptidi non conformati stericamente è trascurabile per quanto riguarda la cinetica di aggregazione. La seconda evidenza è che in alcuni casi GroEL attua il recupero, nella via di folding, di intermedi che ne sono usciti e che si trovano stabilmente in una situazione che non evolve né verso il folding né verso l'aggregazione irreversibile. In questo modo, la chaperonina accresce significativamente la velocità di folding. È probabile che il ruolo attivo delle chaperonine sia legato alla loro funzione di 'srotolamento' di intermedi inclini all'aggregazione o cineticamente intrappolati.
10. La cooperazione tra diverse chaperon durante il folding delle proteine
Proteine appartenenti a differenti classi di chaperon si trovano insieme negli stessi compartimenti cellulari. Esse sono apparentemente non correlate l'una all'altra, hanno un'ampia specificità di substrato e potrebbero assistere il folding di proteine simili. In accordo con l'idea di una ridondanza delle chaperon, alcune di esse risultano non essenziali per la cellula. Per esempio, mutazioni di DnaK non sono letali alle normali temperature; sembra che la perdita della funzione di questa chaperon possa essere compensata dall'espressione di un'altra proteina non correlata con DnaK, ma con funzione sovrapponibile. A temperature elevate DnaK diviene essenziale per la vita, suggerendo quindi che sia necessario l'intero e completo potenziale di tutte le chaperon della cellula. Altre chaperon sono invece essenziali a tutte le temperature (per es., GroEL di E. coli, Hsp60 e Hsp70 della matrice mitocondriale). Ciò suggerisce che queste ultime chaperon svolgano un ruolo insostituibile, che non può essere assunto da un'altra chaperon.
Le chaperonine sono, comunque, coinvolte nella prevenzione dell'aggregazione o nell'assistenza al processo di folding delle proteine. Sembra che esse abbiano un ruolo in un passaggio essenziale di questo processo, che non può essere svolto da un altro tipo di chaperon. La specificità di substrato di diverse classi di chaperon dovrebbe essere, almeno in parte, differente. Effettivamente, mentre le chaperon della classe Hsp70 interagiscono principalmente con proteine completamente denaturate, le chaperonine della classe Hsp60 potrebbero interagire con elementi della struttura secondaria e con intermedi della via di folding; esse infatti legano le strutture globulari semisolide. A differenza delle Hsp70, le chaperonine hanno alta affinità per i peptidi corti e i polipeptidi in conformazione estesa. Queste due classi di chaperon potrebbero, quindi, agire in sequenza nel folding di proteine neosintetizzate o appena traslocate.
bibliografia
Anfinsen 1973: Anfinsen, Christian B., Principles that govern the folding of protein chains, "Science", 181, 1973, pp. 223-230.
Creighton 1990: Creighton, Thomas E., Protein folding, "Biochemical journal", 270, 1990, pp. 1-16.
Creighton 1993: Creighton, Thomas E., Proteins: structures and molecular properties, New York, Freeman, 1993.
Cyr 1994: Cyr, Douglas M. - Langer, Thierry - Douglas, Michael G., DnaJ-like proteins: molecular chaperones and specific regulators of Hsp70, "Trends in biochemical sciences", 19, 1994, pp. 176-181.
Ellis, van der Vies 1991: Ellis, R. John - van der Vies, Saskia M., Molecular chaperones, "Annual review of biochemistry", 60, 1991, pp. 321-347.
Fischer, Schmid 1990: Fischer, Gunter - Schmid, Franz X., The mechanism of protein folding. Implications of in vitro refolding models for de novo protein folding and translocation in the cell, "Biochemistry", 29, 1990, pp. 2205-2212.
Georgopoulos 1992: Georgopoulos, Costa, The emergence of the chaperone machines, "Trends in biochemical sciences", 17, 1992, pp. 295-299.
Gething, Sambrook 1992: Gething, Mary-Jane - Sambrook, Joseph, Protein folding in the cell, "Nature", 355, 1992, pp. 33-45.
Hendrick, Hartl 1993: Hendrick, Joseph P. - Hartl, Franz-Ulrich, Molecular chaperone functions of heat-shock proteins, "Annual review of biochemistry", 62, 1993, pp. 349-384.
Mayhew 1996: Mayhew, Mark e altri, Protein folding in the central cavity of the GroEL-GroES chaperonin complex, "Nature", 379, 1996, pp. 420-426.
McCarty 1995: McCarty, John S. e altri, The role of ATP in the functional cycle of the DnaK chaperone system, "Journal of molecular biology", 249, 1995, pp. 126-137.
Morimoto 1994: The biology of heat shock proteins and molecular chaperones, edited by Richard I. Morimoto, Alfred Tissières, Costa Georgopoulos, Plainview (N.Y.), Cold Spring Harbor Laboratory Press, 1994.
Pfanner, Meijer 1995: Pfanner, Nikolaus - Meijer, Michel, Protein sorting. Pulling in the proteins, "Current biology", 5, 1995, pp. 132-135.
Randall, Hardy 1995: Randall, Linda L. - Hardy, Simon J.S., High selectivity with low specificity: how SecB has solved the paradox of chaperone binding, "Trends in biochemical sciences", 20, 1995, pp. 65-69.
Todd 1994: Todd, Michael J. - Viitanen, Paul V. - Lorimer, George H., Dynamics of the chaperonin ATPase cycle: implications for facilitated protein folding, "Science", 265, 1994, pp. 659-666.
Weissman 1995: Weissman, Jonathan S. e altri, Mechanism of GroEL action: productive release of polypeptide from a sequestered position under GroES, "Cell", 83, 1995, pp. 577-587.
Weissman 1995: Weissman, Jonathan S., All roads lead to Rome? The multiple pathways of protein folding, "Chemistry and biology", 2, 1995, pp. 255-260.