
Microbiologia
Microbiologia
La genomica, che esplora la biologia degli organismi per mezzo del loro progetto genetico, ha profondamente influenzato la disciplina della microbiologia. Essa ha condotto alla revisione delle definizioni di entità microbiche, a riconsiderare le loro capacità e quindi l'insieme di metodi e approcci della microbiologia. Data la vastità della sua influenza sulle diverse sottodiscipline della microbiologia (per es., lo studio del metabolismo, la fisiologia, l'ecologia, le relazioni ospite-patogeno e la microbiologia industriale) e sulle interazioni con le altre discipline (per es., la medicina umana e veterinaria, l'agricoltura, la biologia evoluzionista e la biologia strutturale), l'impatto della genomica sulla microbiologia è stato probabilmente senza pari nell'ultimo secolo.
Recenti studi genomici hanno gettato una nuova e interessante luce sulla nostra suddivisione del mondo microbico in quattro gruppi: Bacteria, Archaea, Eucaria e virus. Alcune delle distinzioni ottenute grazie a metodi ultrastrutturali e biochimici, che per molti anni hanno tenuto separati questi gruppi ‒ per esempio, il livello generale di complessità cellulare, la dimensione della cellula, la grandezza del genoma, la composizione dei ribosomi (se presenti), il tipo di lipidi e la complessità di comportamento ‒ hanno iniziato a sgretolarsi già nell'era precedente alla genomica. Ma la genomica ha amplificato l'effetto e ha reso possibile una comprensione più profonda delle regole e delle eccezioni che caratterizzano i quattro gruppi microbici.
I concetti chiave della microbiologia sono quindi andati incontro a profonde revisioni, proprio in conseguenza dei nuovi strumenti offerti dallo studio dei genomi. Anche il concetto di genoma minimo universale viene messo alla prova e sono in corso ricerche sulla capacità dei genomi minimi di sostenere la complessità cellulare. Vi sono state inoltre interessanti scoperte nello studio delle relazioni interne alle comunità microbiche (la cd. metagenomica): per esempio sono stati osservati l'emergere di strategie cooperative come mediazione di conflitto tra diversi ceppi di batteriofagi in competizione e quindi la rapida evoluzione di meccanismi imprevisti di sopravvivenza. Proprio la possibilità di studiare le comunità microbiche nella loro interezza, senza la necessità di isolare colonie omogenee composte da un unico microrganismo, ha ulteriormente stimolato lo sviluppo di migliori tecniche di coltura.
Infine, sta emergendo il potenziale tassonomico della genomica, poiché gli studi di diversi ceppi ci permettono di rivedere e raffinare il concetto di specie batterica nonché l'idea di genoma statico. Molti degli studi compiuti negli ultimi anni hanno preso in considerazione i genomi di dimensioni estreme ‒ molto grandi e molto piccoli ‒ evidenziandone le diverse proprietà codificanti e studiandone la composizione anche in base ai prodotti proteici che questi genomi sono in grado di esprimere. Così è venuto alla luce che alcuni funghi dotati di genomi estremamente ridotti contengono elementi di metabolismo batterico, mentre in alcuni microrganismi è stata ritrovata una sorta di firma proteica degli eucarioti. Queste scoperte hanno di conseguenza reso più sfumate le distinzioni tra i diversi domini microbici.
Le dimensioni dei genomi
Dallo studio della grandezza dei genomi microbici sono emersi risultati estremamente importanti che hanno permesso di comprendere meglio anche l'evoluzione della vita e in particolare quella dei microrganismi. Un buon esempio viene fornito da recenti analisi dei virus a DNA. Il mimivirus è un virus a DNA che infetta le amebe. È una particella estremamente grande (400 nm) gram-positiva e ha un genoma di 1,2 Mb (megabasi). Il genoma contiene geni finora ignoti nei virus, che codificano per proteine coinvolte nella traduzione delle proteine e dei componenti dei pathway metabolici. Per la dimensione cellulare, la grandezza del genoma e il contenuto genico, questo mimivirus imita un piccolo batterio. Recentemente è stato sostenuto che l'espansione della definizione di virus in seguito alla scoperta dei mimivirus è di natura quantitativa e non qualitativa. Sarebbe invece qualitativa la diversità presentata dal genoma del virus polyDNA Cotesia congregata bracovirus (CcBv). Il genoma del mimivirus codifica per proteine coinvolte nella trascrizione, traduzione e replicazione del DNA; pochissime di queste si trovano nel genoma del CcBv. Inoltre, molti geni virali tipici sono assenti dal genoma del CcBv. L'affascinante ciclo biologico del CcBv è strettamente associato a due specie eucariotiche: le vespe parassite Braconidi e il bruco che esse predano. L'espressione dei geni virali nel bruco ostacola la risposta immunitaria e aiuta la sopravvivenza della vespa parassita. L'analisi genomica di CcBv ha rivelato peculiarità come la densità di codificazione estremamente bassa, l'assenza di geni deputati alla codifica di proteine per la replicazione del DNA e l'alta percentuale (70%) di geni che sembrano contenere introni. Tutte insieme, queste caratteristiche somigliano a un segmento di DNA di genoma eucariotico piuttosto che a un genoma virale.
Tuttavia, questi modelli genici non sono ancora stati testati in vitro (per es., per mezzo dell'analisi delle sequenze di cDNA) e quindi il loro significato non può ancora essere pienamente valutato.
La dimensione e il contenuto del genoma
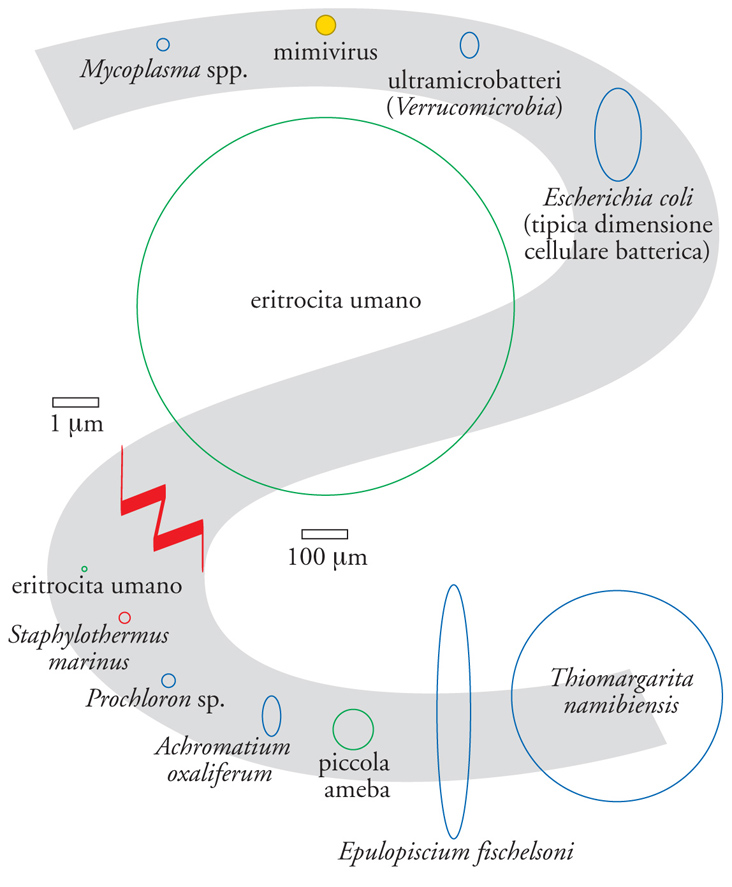
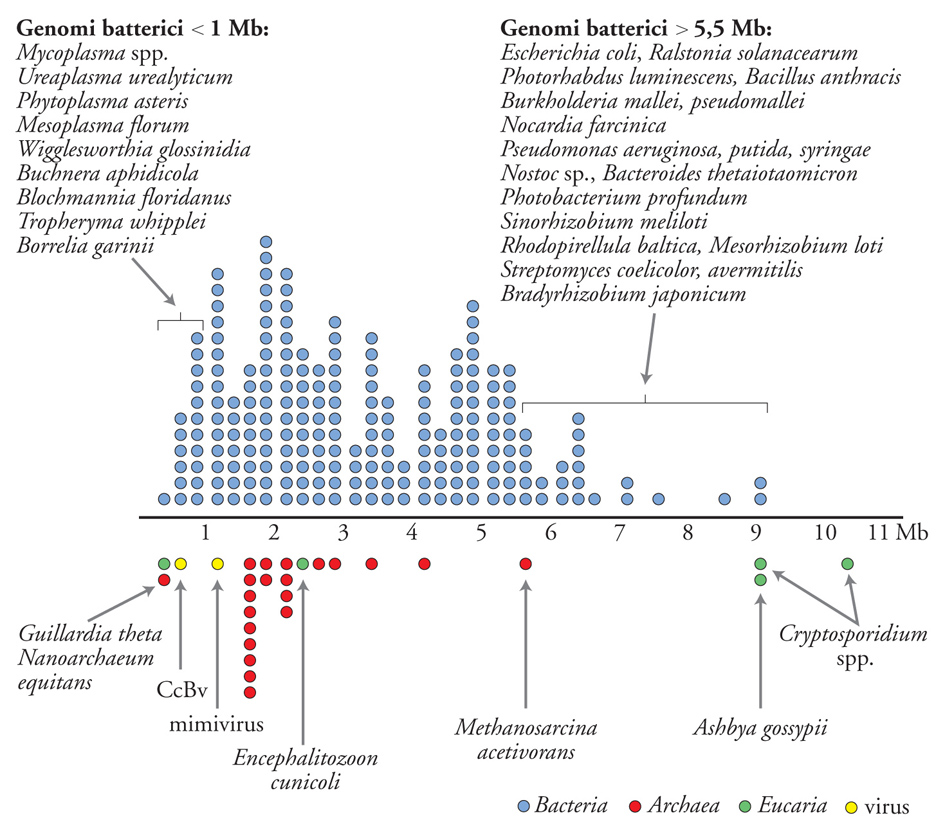
Lo spostamento dei confini tra i domini microbiologici fa seguito a risultati emersi negli ultimi quindici anni che a loro volta avevano complicato le definizioni microbiologiche. Relativamente alla dimensione cellulare, queste ultime comprendevano la scoperta di cellule batteriche molto grandi, come Epulopiscium, e molto piccole, come gli ultramicrobatteri e Nanoarchaeum (fig. 2). A ciò si è aggiunta l'osservazione che esistono genomi batterici più piccoli di quelli del mimivirus e più grandi di alcuni genomi fungini (fig. 3).
In un'analisi del contenuto funzionale di questi grandi genomi procarioti, è stata dimostrata una sovrarappresentazione di alcune categorie di geni, come quelli per la regolazione della trascrizione e del metabolismo secondario. Al contrario, altre categorie di geni, come quelli per la traduzione e l'elaborazione del DNA, sono relativamente meno presenti nei grandi genomi. La mancanza di correlazione con i genomi non codificanti e un ipotetico contesto aperto di lettura dei genomi hanno portato alla conclusione che l'espansione del genoma favorisce specifiche classi funzionali. Alcuni gruppi filogenetici sono sovrarappresentati in queste analisi, quindi sarà interessante vedere se queste tendenze verranno confermate quando i genomi conosciuti saranno maggiormente diversificati.
Analisi recenti di diversi funghi hanno rivelato che essi hanno genomi efficienti di minima dimensione, alcuni dei quali hanno adottato elementi del metabolismo batterico. Quando fu pubblicato il genoma del fungo filamentoso Ashbya gossypii nel 2004, rappresentava il più piccolo eucariota autonomo noto (9,2 Mb). È un genoma molto compatto, con breve distanza tra i geni e solo 221 introni. Il suo alto livello di omologia e di sintenia con il genoma di Saccharomyces cerevisiae (il lievito di birra) e la presenza di un numero simile di proteine indicano che A. gossypii rappresenta la dimensione minima del genoma per un fungo autosufficiente e che l'evoluzione di S. cerevisiae ha implicato la duplicazione o la fusione di due specie imparentate. Anche due genomi di Cryptosporidium mostrano pathway metabolici molto semplificati, che si basano sulla glicolisi per la produzione di energia. Essi sono particolarmente dotati di enzimi che mostrano molte somiglianze con le controparti batteriche, tra cui enzimi di conversione dei nucleotidi, sintasi degli acidi grassi e sintasi di polichetidi. Ancora, era stato previsto che il genoma di Neurospora crassa, molto più grande (40 Mb), codificasse anche elementi della fotobiologia a luce rossa, tra cui due presunti omologhi fitocromi che presentano grande somiglianza con i fitocromi batterici.
L'acquisizione da parte degli Eucaria dei componenti metabolici dei batteri non sembra essere limitata ai funghi. Il genoma ‒ da poco pubblicato ‒ di Entamoeba histolytica, così come gli altri lignaggi di protisti amitocondriali, rappresentati da Giardia e Thricomonas, sembra avere ridotto o forse eliminato gran parte dei pathway metabolici mitocondriali e utilizzato pathway anaerobici associabili ai procarioti anaerobici. Sembra quindi possibile ipotizzare che il trasferimento orizzontale di 96 geni dai batteri sia stato il più probabile meccanismo di acquisizione di enzimi di fermentazione e di trasportatori del glucosio simil-batterici, nonché delle proteine per la resistenza agli stress ossidativo e nitrosativo.
Su una scala più globale, il genoma di 13,8 Mb di Schyzosaccharomyces pombe ha reso possibile uno sguardo alla transizione dai procarioti agli eucarioti, che secondo i calcoli dovrebbe aver richiesto un maggior numero di nuovi geni rispetto al passaggio da procariote a eucariote. È stata quindi avanzata l'ipotesi che la distinzione tra eucariote e procariote non dipenda solo dal numero di geni, ma anche dal loro tipo e dalla regolazione. I genomi dei protisti, come quello del parassita della malaria Plasmodium falciparum, hanno inoltre aiutato a definire cosa caratterizza un eucariote. Tuttavia, alcuni procarioti mostrano una chiara affinità con alcune proteine esclusive degli Eucaria e degli Archaea. Per esempio le batteriorodopsine (le pompe di protoni fotodipendenti) prima erano note solamente negli archei alofili, ma sono state ritrovate in un gammaproteobatterio del plancton. Un altro esempio è rappresentato dalle tubuline, proteine che costituiscono i microtubuli del citoscheletro degli eucarioti, la cui presenza era prevista nella sequenza genomica di Prosthecobacter dejongeii. L'assemblaggio in vitro e l'idrolisi del GTP (guanosina trifosfato) sono stati dimostrati per queste proteine. Le tubuline erano prima ignote nei batteri e negli archei. Su 347 proteine tipiche degli eucarioti, 10 sono state identificate nel genoma di Prosthecobacter dejongeii (phylum Verrucomicrobia) e 17 nel genoma di Gemmata sp. Wa-1 (phylum Planctomycetes). Ciò suggerisce che membri ancestrali di questi phyla potrebbero aver dato origine a un organismo proto-eucariote.
La natura del genoma minimo
Le riflessioni sulla dimensione del genoma hanno naturalmente portato alle questioni riguardanti la ridondanza del genoma, ovvero, quanta parte del genoma è assolutamente necessaria alla vita? Uno studio recente di mutagenesi ad alta densità suggerisce che i requisiti essenziali strutturali e metabolici di Mycoplasma tuberculosis sono abbastanza differenti da quelli di Haemophilus influenzae. Alcune di queste differenze hanno un senso nel contesto del peculiare tipo di parete cellulare dei micobatteri (per es., la presenza di meccanismi per il metabolismo dei lipidi e del ramnosio in M. tuberculosis). Nei genomi degli endosimbionti degli afidi, il genoma minimo teorico non riflette il complemento genetico di alcuna cellula che si ritrova in natura. Questi risultati mettono in discussione il concetto di genoma minimo universale e hanno portato all'idea che i geni ortologhi condivisi non definiscano l'insieme minimo di geni.
Anche la capacità di organismi genomicamente limitati di produrre strutture cellulari relativamente complesse è in corso di analisi. Mycoplasma pneumoniae sintetizza un organello terminale che si lega all'epitelio dell'ospite. La natura polare di questa struttura e il possesso di elementi citoscheletrici sollevano interessanti quesiti su come vengano effettuate la replicazione e la migrazione interna alla cellula, utilizzando uno scarso complemento genico.
Le relazioni tra microrganismie le loro comunità
Studi recenti hanno esteso il campo della genomica della simbiosi, prima stabilmente posizionata nei domini batterici e degli Eucaria, agli Archaea e ai virus. Il genoma dell'archea Nanoarchaeum equitans (un parassita di un altro archeo) ha il più piccolo genoma procariota completo (di soli 490 kb) finora noto. Sembra che questo genoma sia andato incontro a una riduzione evolutiva così come è accaduto per i parassiti batterici. Il genoma sembra essere relativamente stabile, senza pseudogeni o regioni non codificanti. Da questi indizi, si può sostenere che la divergenza da altre linee evolutive di archea sia stata un evento molto antico.
Sono state inoltre individuate relazioni microbiche meno amichevoli, per esempio il genoma di Bdellovibrio bacteriovirus, che fa da predatore su batteri gram-negativi con attacchi rigidamente coordinati. Uno studio sui batteriofagi di Escherichia coli ha consentito di analizzare le interazioni tra microrganismi allo scopo di adattarsi a due pressioni selettive opposte: cooperazione e competizione. I fagi sono stati indotti ad adottare un ciclo cellulare che comprende stadi sia di conflitto che di cooperazione. Ciò è stato possibile ingegnerizzando ogni fago con geni di diverse resistenze ad antibiotici e, quindi, spingendoli alla crescita sotto la selezione di entrambi gli antibiotici, per favorire l'evoluzione di una rapida risoluzione dei conflitti. In breve tempo, è stata raggiunta una mediazione, con uno dei fagi che ha drasticamente ridotto la dimensione del suo genoma perdendo i geni che codificano il virione ed entrambi i fagi che sono stati impacchettati dalla stessa capsula proteica. Questo impacchettamento in comune lega gli interessi adattativi dei fagi; la prova sperimentale di questo meccanismo di risoluzione dei conflitti ha dimostrato che la cooperazione, necessaria per la transizione degli organismi a un livello superiore di complessità biologica, può evolvere rapidamente.
Coltivare o non coltivare?
La genomica ci ha portato a rivedere non solo i concetti di cos'è un microrganismo e di cosa fa, ma anche il miglior approccio per studiare questi organismi. La coltura dei microrganismi in colture pure, che è stata per decenni l'unica strada per l'identificazione e la caratterizzazione microbica, è stata eclissata dalla nuova metodologia indipendente dalla coltura, che ha preso piede negli anni Novanta del Novecento. Questo approccio, che ha analizzato principalmente i geni delle piccole subunità dell'RNA ribosomale in popolazioni miste di microrganismi provenienti da campioni ambientali, ha rivelato la diversità del mondo procariota mai messo in coltura prima, fornendo la base per i primi studi di biogeografia microbica. Per quanto questo campo fosse interessante, di fatto fornì solo un insieme di etichette per i membri di una comunità microbica, senza indicazioni sulle loro attività e quindi sul loro ruolo ecologico in quella comunità (con l'eccezione di alcuni taxa per i quali la funzione poteva essere estrapolata dalla tassonomia). La previsione funzionale necessitava dell'accesso al genoma piuttosto che al singolo gene e lo sviluppo delle tecnologie di sequenziamento ad alta efficienza hanno consentito tale accesso. Ciò ha dato vita alla metagenomica (nota anche come genomica delle comunità, ecogenomica o genomica ambientale), l'analisi genomica diretta delle comunità microbiche che abitano un campione ambientale, senza dover separare un organismo dal suo habitat e mantenerlo in un mezzo di coltura su substrati artificiali.
Un effetto collaterale dell'analisi metagenomica è stato la consapevolezza della difficoltà di assemblare un genoma microbico completo a partire dai dati metagenomici, con l'eccezione degli studi che hanno preso in considerazione campioni a bassa complessità, e delle istituzioni che hanno le risorse per generare una grande mole di dati delle sequenze. Anche a causa di questa attuale limitazione, i microbiologi stanno mostrando un rinnovato interesse nell'arte della coltura microbica. L'esempio più importante è la coltivazione di SAR11, un clado dominante e ubiquo di eterotrofi marini, che per molti anni si era dimostrato recalcitrante alla coltura. Si è riusciti in questa impresa per mezzo di una combinazione di tecniche di coltura ad alta efficienza (piastre con microvolumi) utilizzando un mezzo diluito, e uno screening rapido e sensibile con l'impiego di sonde fluorescenti specifiche per il gruppo SAR11. La disponibilità di questi microrganismi, una volta isolati permetterà di sequenziarne il genoma e quindi la ricerca sulle basi genomiche della loro biologia. Ciò riveste un particolare interesse a causa della ridottissima dimensione cellulare (0,1 μm3) di questi organismi autosufficienti. Questo primo successo è stato poi seguito da molti altri, che hanno isolato membri di gruppi microbici ubiqui finora poco coltivati, per mezzo di manipolazioni delle condizioni di coltura, anche solo grazie a semplici aggiustamenti come l'uso di formulazioni solide piuttosto che liquide dello stesso mezzo di coltura o l'aumento del tempo di incubazione.
La genomica potrebbe quindi continuare a essere l'unica metodologia molecolare che ha facilitato o incoraggiato lo sviluppo sia degli approcci microbiologici legati alle colture sia di quelli indipendenti da esse, approcci che erano considerati strategie rivali piuttosto che complementari. Mentre i dati genomici si continuano ad accumulare, sia dalle colture che dai campioni ambientali, sarà interessante vedere come gli approcci si influenzeranno a vicenda.
Che cos'è una specie batterica?
Questa domanda ha per anni afflitto i microbiologi e ha confuso i biologi degli organismi superiori, a causa della scarsità di caratteri fenotipici utili per la tassonomia, dell'incapacità della principale molecola tassonomica (rRNA) di distinguere gruppi molto correlati e della laboriosità e mancanza di riproducibilità dell'ibridazione DNA-DNA. La crescente disponibilità di dati genomici da diversi ceppi di una specie promette di fornire una misura quantitativa della somiglianza tra genomi e quindi dello status di specie. All'altro estremo dello spettro tassonomico, l'analisi dei membri rappresentativi di phyla poco conosciuti promette di chiarire la reale diversità dei genomi microbici e ci permetterà di comprendere meglio le relazioni filogenetiche tra i phyla. L'interfaccia tra la sistematica dei microrganismi, l'evoluzione e la genomica darà inoltre l'opportunità di riconsiderare il genoma come entità statica e, particolarmente, la sua suscettibilità ad acquisire o perdere geni per trasferimento genetico laterale all'interno e all'esterno dei confini di specie.
L'analisi filogenetica di diversi ceppi di Staphylococcus aureus ha mostrato che la diversificazione della regione ad alta variabilità RD13, che codifica proteine forse correlate alla patogenicità, ha probabilmente avuto luogo per mezzo del trasferimento genico laterale e per ricombinazione. Sembra inoltre che i ceppi di Thermotoga maritima spesso scambino geni con ceppi di Th. neapolitana. Secondo alcuni studiosi, tali scambi avvengono anche tra entità filogenetiche ancora più distanti (fino ai phyla) e perfino tra i batteri e i funghi o i protisti. Altri autori hanno invece invitato alla prudenza nell'uso dei fattori composizionali per determinare l'origine filogenetica dei geni trasferiti lateralmente, sostenendo che questi fattori non riflettono il contesto genomico della fonte originale, ma piuttosto un'associazione con elementi mobili, quali i fagi.
L'impatto relativo del trasferimento genico laterale sull'evoluzione e la diversificazione microbica rimane un tema aspramente dibattuto, al punto da consigliare l'inizio di un processo graduale di accordo interno alla comunità.
È probabile che con l'accumulo di maggiori dati relativi ai genomi, sia da isolati coltivati sia da fonti metagenomiche, continueremo a scoprire affascinanti eccezioni alle regole che governano l'identità e le capacità dei microrganismi. Ci si possono aspettare relazioni più complesse e interessanti tra i quattro domini microbici (Bacteria, Archaea, Eucaria e i virus), ma anche in altri contesti, come le divisioni autotrofi-eterotrofi, aerobici-anaerobici, autosufficienti-simbionti, che potrebbero diventare sempre più simili a spettri piuttosto che a dicotomie. Possiamo anche guardare con ottimismo allo sviluppo di una tassonomia basata sul genoma, maggiormente quantitativa e rigorosa di quanto possa essere oggi con i nostri limitati cronometri molecolari e tratti fenotipici. Senza alcun dubbio, la genomica continuerà a fornire imprevedibili sfide e sviluppi ai concetti della microbiologia.
Bibliografia
Angert 1993: Angert, Esther R. - Clements, Kendall D. - Pace, Norman R., The largest bacterium, "Nature", 362, 1993, pp. 239-241.
Daubin 2003: Daubin, Vincent - Lerat, Emmanuelle - Perrière, Guy, The source of laterally transferred genes in bacterial genomes, "Genome biology", 4, 2003, p. R57.
Espagne 2004: Espagne, Eric, Genome sequence of a polydnavirus: insights into symbiotic virus evolution, "Science", 306, 2004, pp. 286-289.
Janssen 1997: Janssen, Peter H. e altri, Novel anaerobic ultramicrobacteria belonging to the Verrucomicrobiales lineage of bacterial descent isolated by dilution culture from anoxic rice paddy soil, "Applied and environmental microbiology", 63, 1997, pp. 1382-1388.
Konstantinidis, Tiedje 2005: Konstantinidis, Konstantinos T. - Tiedje, James M., Genomic insights that advance the species definition for prokaryotes, "Proceedings of the National Academy of Sciences USA", 102, 2005, pp. 2567-2572.
Raoult 2004: Raoult, Didier e altri, The 1.2-Mb genome sequence of mimivirus, "Science", 306, 2004, pp. 1344-1350.
Riesenfeld 2004: Riesenfeld, Christian S. - Schloss, Patrick D. - Handelsman, Jo, Metagenomics: genomic analysis of microbial communities, "Annual review of genetics", 38, 2004, pp. 525-552.
Sachs, Bull 2005: Sachs, Joel L. - Bull, James J., Experimental evolution of conflict mediation between genomes, "Proceedings of the National Academy of Sciences USA", 102, 2005, pp. 390-395.
Sait 2002: Sait, Michelle - Hugenholtz, Philip - Janssen, Peter H., Cultivation of globally distributed soil bacteria from phylogenetic lineages previously only detected in cultivation-independent surveys, "Environmental microbiology", 4, 2002, pp. 654-666.
Stackebrandt 2002: Stackebrandt, Erko e altri, Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology, "International journal of systematic and evolutionary microbiology", 52, 2002, pp. 1043-1047.
Tyson 2004: Tyson, Gene W. e altri, Community structure and metabolism through reconstruction of microbial genomes from the environment, "Nature", 428, 2004, pp. 37-43.
Venter 2004: Venter, J. Craig e altri, Environmental genome shotgun sequencing of the Sargasso Sea, "Science", 304, 2004, pp. 66-74.