
Diabete
Diabete
Dal greco diabaíno ('passo attraverso'), il termine 'diabete' indica due entità cliniche assai distinte, ma accumunate dal passaggio attraverso i reni di un eccesso di urina (poliuria): il diabete mellito (dal latino mel = miele, per il sapore dolciastro delle urine) e il diabete insipido.
Il diabete mellito, o semplicemente diabete, è causato da una carenza, assoluta o relativa, di insulina con conseguente iperglicemia (aumento delle concentrazioni plasmatiche di glucosio). I sintomi dell'iperglicemia includono la poliuria (aumento della quantità di urina emessa nelle 24 ore), la conseguente polidipsia (sete intensa), calo ponderale e polifagia. Si associano inoltre una suscettibilità alle infezioni e, nell'età evolutiva, un deficit di accrescimento. Le complicanze acute della grave iperglicemia sono rappresentate dalla chetoacidosi e dalla sindrome iperosmolare non chetotica, che possono condurre al coma e all'exitus. L'iperglicemia cronica provoca negli anni alterazioni strutturali e funzionali di vari organi, specie occhi (retinopatia) con rischio di cecità, reni (nefropatia) con rischio di insufficienza renale, nervi (neuropatia) con rischio di impotenza coeundi e di ulcere podaliche che possono richiedere l'amputazione, soprattutto quando coesiste una arteriopatia ostruttiva degli arti inferiori per accelerata progressione di aterosclerosi; quest'ultima condiziona anche un aumentato rischio di eventi cardio- (infarto) e cerebro-vascolari (ictus).
La maggior parte dei pazienti è affetta da uno dei due sottotipi principali della malattia: il 'diabete tipo 1', con esordio durante l'infanzia o l'adolescenza, è caratterizzato da un deficit assoluto di secrezione insulinica che richiede, per la sopravvivenza, la somministrazione di insulina esogena, da cui la definizione precedentemente utilizzata di 'diabete insulino-dipendente'; e il 'diabete tipo 2' (in passato definito 'non insulino-dipendente'), generalmente con esordio nell'età adulta ed elevata prevalenza (ca. 4%) nei Paesi industrializzati; esso è caratterizzato da resistenza all'azione dell'insulina (insulino-resistenza) e da un inadeguato aumento compensatorio della secrezione insulinica.
La terapia del diabete tipo 1 si basa sulla somministrazione giornaliera di multiple iniezioni sottocutanee di insulina. Il trapianto di pancreas, a causa della necessità di seguire per tutta la vita una terapia antirigetto più dannosa del diabete stesso, è indicato solo in associazione al trapianto di rene. La terapia iniziale del diabete tipo 2 si basa su dieta ed esercizio fisico. Successivamente vengono utilizzati farmaci, definiti 'antidiabetici orali', che appartengono a tre categorie principali: insulino-sensibilizzanti, secretagoghi (che stimolano la secrezione insulinica) e inibitori dell'assorbimento degli zuccheri a livello intestinale. Nelle fasi più avanzate della malattia può essere necessario associare una o più somministrazioni di insulina.
Il diabete insipido è una malattia relativamente rara caratterizzata da poliuria (da 2,5 l nei casi lievi fino a 20 l nelle 24 ore nei casi gravi) per incapacità del rene di concentrare le urine e da conseguente polidipsia compensatoria. I segni di disidratazione sono rari, a meno che l'assunzione di liquidi sia impedita o il paziente non avverta lo stimolo della sete (stati di incoscienza). L'eziopatogenesi è legata a un difetto di produzione o di azione dell'ormone antidiuretico.
Diabete mellito
Cenni di fisiologia dell'omeostasi glicidica
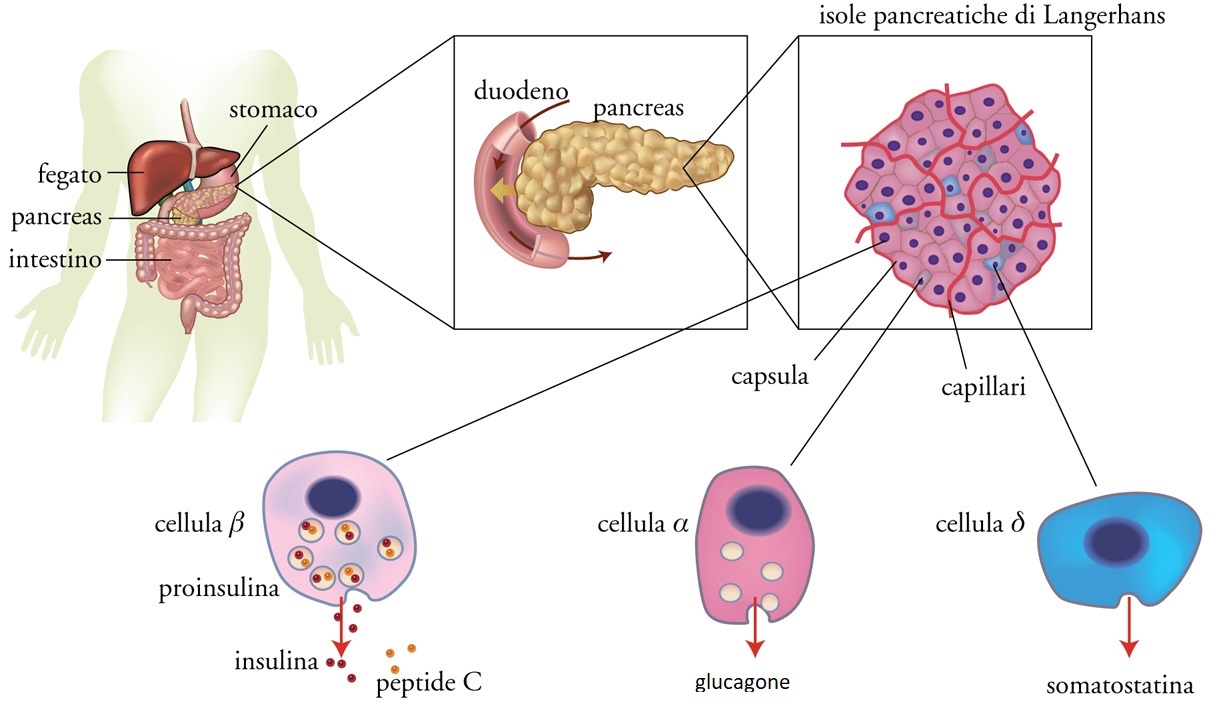
Il mantenimento di una normale omeostasi glicidica (che consente all'organismo di disporre costantemente del giusto quantitativo di glucosio, suo principale substrato energetico) è il risultato della fine orchestrazione di tre processi fondamentali: la secrezione insulinica stimolata dall'assorbimento del glucosio a livello intestinale, la produzione di glucosio da parte del fegato e la sua utilizzazione da parte di quasi tutti i tessuti dell'organismo; tali eventi sono differentemente modulati in relazione alle diverse fasi del ciclo alimentazione-digiuno. L'insulina è un ormone i cui effetti conducono in ultima analisi a una aumentata sintesi di glicogeno (forma sotto la quale il glucosio viene immagazzinato nel fegato e nel muscolo scheletrico), lipidi (nel tessuto adiposo) e proteine (nel muscolo scheletrico); essa è prodotta dalle cellule β delle isole pancreatiche di Langerhans in risposta ad aumenti della glicemia (fig. 2).
Metabolismo del glucosio nelle fasi di digiuno. - Da sei a dodici ore dopo un pasto avviene la transizione dalla fase postprandiale alla fase di digiuno. Il glucosio che entra nella circolazione viene prodotto dal fegato e, se il digiuno viene prolungato per alcuni giorni, anche dal rene. Il glucosio epatico deriva sia dalla scissione del glicogeno, preponderante dopo il digiuno notturno, sia dalla sintesi di nuove molecole di glucosio: tale processo, definito 'neoglucogenesi', si attiva dopo l'esaurimento delle scorte di glicogeno quando il digiuno viene prolungato. Circa l'80% della captazione di glucosio avviene in modo insulino-indipendente nel cervello, nei visceri addominali e nei globuli rossi. Il muscolo, al contrario, è il tessuto insulino-sensibile per eccellenza ed è responsabile per gran parte della utilizzazione postprandiale del glucosio. Esso capta poco glucosio durante il digiuno, perché i livelli plasmatici di insulina sono bassi, e utilizza principalmente gli acidi grassi liberi, così come fegato, cuore e rene.
Metabolismo postprandiale del glucosio. - L'incremento postprandiale della glicemia stimola la secrezione insulinica da parte delle cellule β del pancreas endocrino. La captazione postprandiale di glucosio da parte del fegato permette di ripristinare le scorte di glicogeno utilizzate nella precedente fase di digiuno. La captazione muscolare di glucosio, che durante il digiuno avviene solo in minima parte, diviene preponderante rispetto agli altri tessuti.
Classificazione
L'attuale classificazione prevede la suddivisione del diabete in quattro gruppi; i primi due gruppi si identificano rispettivamente con il diabete tipo 1 e il diabete tipo 2 che sono di gran lunga i più comuni. Il primo è causato da una distruzione, autoimmunitaria o idiopatica, delle cellule β del pancreas endocrino; il secondo è originato da un deficit combinato di secrezione e azione insulinica. Il terzo gruppo include forme genetiche causate da mutazioni di singoli geni con trasmissione ereditaria di tipo monogenico, che codificano per proteine coinvolte nella secrezione insulinica (indicate con l'acronimo inglese MODY, Maturityonset diabetes of the young, per le caratteristiche cliniche assimilabili al diabete dell'adulto ma con insorgenza in età giovanile) o nella sua azione a livello dei tessuti bersaglio (sindromi da estrema insulino-resistenza); forme secondarie causate da malattie del pancreas esocrino (pancreatiti, traumi o neoplasie del pancreas, ecc.) e da altre endocrinopatie (acromegalia, malattia di Cushing, glucagonoma, feocromocitoma, ipertiroidismo, somatostatinoma, aldosteronoma ecc.), da assunzione di alcuni farmaci (cortisonici, diazossido, diuretici tiazidici, interferone ecc.), da alcune infezioni (rosolia congenita, citomegalovirus ecc.); altre forme presenti in quadri sindromici specifici (sindrome di Down, sindrome di Laurence-Moon-Biedl, sindrome di Prader-Willi ecc.). Il quarto gruppo include infine il diabete gestazionale, che si sviluppa durante la gravidanza in donne che abbiano familiarità per diabete e/o che presentino fattori di rischio quali sovrappeso od obesità.
Diagnosi
Viene posta diagnosi di diabete qualora uno dei seguenti tre criteri venga soddisfatto: (a) sintomi tipici di diabete, associati a una glicemia rilevata in qualsiasi momento della giornata, indipendentemente dall'assunzione o meno di cibo, uguale a o maggiore di 200 mg/dl; (b) glicemia misurata dopo almeno otto ore di digiuno uguale a o maggiore di 126 mg/dl; (c) glicemia dopo due ore dall'assunzione orale di 75 g di glucosio (carico orale di glucosio) uguale a o maggiore di 200 mg/dl. La misurazione della glicemia a digiuno e dopo carico orale di glucosio consente anche di identificare le seguenti categorie intermedie che possono precedere il diabete conclamato (prediabete) e rappresentano quindi campanelli d'allarme: (a) alterata glicemia a digiuno (IFG, Impaired fasting glucose), con valori di glicemia a digiuno compresi tra 100 e 125 mg/dl; (b) intolleranza ai carboidrati (IGT, Impaired glucose tolerance), con valori di glicemia dopo due ore da un carico orale di glucosio compresi tra 140 e 200 mg/dl.
Diabete tipo 1
Il diabete tipo 1 (DT1), che rappresenta circa il 5-10% di tutte le forme di diabete, è una malattia autoimmune caratterizzata dalla progressiva e selettiva distruzione delle cellule β‚ producenti insulina, delle isole pancreatiche, con conseguente carenza insulinica e iperglicemia che, se non corretta con adeguata terapia insulinica, conduce al coma chetoacidosico e all'exitus (da cui la precedente definizione di 'diabete insulino-dipendente'). In oltre il 90% dei casi la malattia esordisce in età infanto-giovanile (⟨20 anni), il che giustifica la definizione di diabete giovanile delle classificazioni iniziali; tuttavia, il DT1 può svilupparsi a qualsiasi età e vengono descritte forme a lenta insorgenza che in fase iniziale non necessitano di insulina (LADA, Late autoimmune diabetes in adults, oppure NIRAD, Non insulin required autoimmune diabetes). Nonostante l'esordio tipico della malattia sia acuto, la distruzione delle cellule β avviene in un periodo di tempo, definito 'preclinico', di durata variabile ma dell'ordine di anni, durante il quale il numero delle cellule insulino-secernenti si riduce progressivamente fino a che la secrezione insulinica diviene insufficiente al mantenimento dell'omeostasi glicidica.
Si ritiene che il processo autoimmune responsabile della distruzione delle cellule β si inneschi in soggetti geneticamente predisposti (per es., portatori di polimorfismi di geni del sistema HLA, Human leucocyte antigens, che modulano la risposta immunitaria) probabilmente in seguito all'intervento di uno o più fattori ambientali precipitanti (virus, tossine, fattori dietetici). A ciò fa seguito l'attivazione del processo autoimmune, determinato dalla perdita della tolleranza immunitaria nei confronti di una o più molecole delle cellule β pancreatiche, con conseguente sviluppo di 'insulite' con infiltrazione di linfociti T che conducono alla distruzione delle cellule β. Marcatori del processo autoimmune sono gli autoanticorpi diretti verso antigeni delle cellule β; essi sono dosabili nel siero dei soggetti affetti e di soggetti a rischio di sviluppare la malattia: anticorpi anti-insula pancreatica (anti-ICA), anticorpi anti-insulina, anticorpi anti-GAD e anti-IA-2. I pazienti con DT1 hanno anche una maggiore suscettibilità ad ammalarsi di altre malattie autoimmuni quali il morbo di Basedow (una forma di ipertiroidismo), la tiroidite di Hashimoto (che evolve lentamente verso l'ipotiroidismo), il morbo di Addison (ipocorticosurrenalismo), la vitiligine e il morbo celiaco.
Diabete tipo 2
Il diabete tipo 2 (DT2) è la forma di diabete più frequente nei Paesi industrializzati (90-95%). La tendenza al costante e progressivo aumento di prevalenza della malattia spinge all'uso sempre più frequente del termine 'epidemia': attualmente sono quasi 150 milioni i diabetici tipo 2 nel mondo e si ritiene che tale numero possa raddoppiare nel 2030. Il DT2 è un'entità clinica assai più sfumata ed eterogenea rispetto al DT1, che nella grande maggioranza dei casi si associa a sovrappeso od obesità e probabilmente riconosce in uno stile di vita caratterizzato da un bilancio energetico positivo (aumento dell'introito calorico in rapporto al dispendio energetico) il suo momento patogenetico fondamentale, sia pure in associazione a una predisposizione genetica. La malattia conclamata è preceduta da un lungo periodo, della durata di decenni, caratterizzato dal progressivo deterioramento dell'omeostasi glicidica causato da una ridotta sensibilità all'insulina a livello dei tessuti bersaglio (muscolo scheletrico, tessuto adiposo, fegato) e da una conseguente ipersecrezione compensatoria dell'ormone, che con il tempo diviene inadeguata, con sviluppo di prediabete (iperglicemia a digiuno e/o intolleranza ai carboidrati) e diabete. Il DT2 va considerato una sindrome più complessa della semplice iperglicemia. Esso infatti si associa in vario grado ad alterazioni lipidiche e aumento della pressione arteriosa, che assieme all'obesità addominale e alle alterazioni dell'omeostasi glicidica costituiscono quella che, con attuale discordanza classificativa, viene definita 'sindrome metabolica': patologia multifattoriale che aumenta il rischio cardiovascolare e alla cui genesi concorrono fattori genetici (a oggi in larga parte ignoti) e ambientali (bilancio energetico positivo per sedentarietà e abbondanza di cibi grassi).
Fisiopatologia. - Da un punto di vista fisiopatologico, l'accumulo delle calorie in eccesso in forma di trigliceridi nel tessuto adiposo viscerale rappresenta probabilmente l'alterazione iniziale alla base delle alterazioni metaboliche che conducono, attraverso la sindrome metabolica, al DT2. A causa della sua posizione anatomicamente strategica, questo deposito adiposo, situato intorno ai visceri all'interno della cavità addominale, riversa gli acidi grassi liberi derivanti dalla più elevata attività lipolitica (scissione dei trigliceridi in glicerolo e acidi grassi) direttamente nel fegato, dove inducono insulino-resistenza con conseguente aumento della produzione epatica di glucosio e tendenza all'iperglicemia a digiuno. Insulino-resistenza e iperafflusso di acidi grassi liberi conducono a steatosi non alcolica (NAFLD, Non alcoholic fatty liver disease, o, volgarmente, 'fegato grasso'). Oltre alla steatosi epatica va detto che l'accumulo di trigliceridi al di fuori del tessuto adiposo riguarda anche il muscolo scheletrico, con conseguente ulteriore riduzione della captazione di glucosio insulino-mediata. Inoltre il tessuto adiposo viscerale, rispetto al grasso sottocutaneo, produce maggiori quantità di molecole ormonali (cioè di sostanze che vengono riversate nel torrente circolatorio e agiscono a distanza in tessuti definiti 'bersaglio') dette 'adipochine', che peggiorano l'insulino-resistenza e inducono un atteggiamento generale pro-infiammatorio. Il quadro fisiopatologico è dominato dall'insulino-resistenza che impone alle cellule β pancreatiche un aumento compensatorio della secrezione insulinica. Iperattività compensatoria, lipotossicità (per aumento degli acidi grassi liberi), infiammazione di basso grado e stress ossidativo (per aumentato metabolismo di grassi e zuccheri) conducono nel tempo a una ridotta riserva funzionale delle cellule β, che si manifesta inizialmente con una ridotta tolleranza glicidica (IGT, Impaired glucose tolerance), per alterazione della prima fase (fase rapida) di secrezione insulinica. A questo punto l'evoluzione verso il diabete subisce un'accelerazione da parte dei sopraggiunti effetti glucotossici che conducono alla fase della malattia conclamata.
I medesimi meccanismi che dall'obesità addominale conducono al diabete concorrono sinergicamente alla progressione della placca ateromasica (quindi all'aterosclerosi con progressivo restringimento delle arterie e rischio di rottura della placca che, se avviene nelle coronarie o nelle carotidi, determina rispettivamente infarto e ictus); è evidente come, nuovamente, fattori genetici e ambientali (tipo di dieta, stress, tabagismo ecc.) possano aggravare o rendere meno manifeste alcune delle diverse componenti, contribuendo così alla eterogeneità fenotipica della sindrome metabolica e quindi del diabete tipo 2. In questo contesto, infine, non si può non menzionare la 'teoria del genotipo economo' per tentare di spiegare l'elevatissima prevalenza della sindrome metabolica e del DT2 nelle società occidentali. Popolazioni un tempo esposte a continue variazioni nell'apporto di cibo (carestia/abbondanza) hanno selezionato un genotipo che ottimizzasse l'accumulo di scorte energetiche in periodi di abbondanza di cibo, permettendo la sopravvivenza in periodi di ridotto apporto calorico. Secondo questa ipotesi, il genotipo economo determinerebbe una selettiva insulino-resistenza a livello muscolare, che permetterebbe al muscolo di non bruciare tutte le energie in periodi di digiuno e di deviarle verso l'accumulo di grasso nei periodi di abbondanza. In questo modo la massa muscolare si preserva più a lungo, aumentando le possibilità di ricerca del cibo. Gli aborigeni australiani, così come i Pima dell'Arizona, rappresentano un esempio classico di questo fenomeno: quando esposti a una dieta occidentale, l'incidenza di diabete aumenta in maniera drammatica, passando dallo 0-1% al 25-40%.
Clinica. - Dal punto di vista clinico l'esordio del DT2 è lento, subdolo e asintomatico nelle fasi iniziali; spesso la diagnosi viene posta in seguito ad analisi emato-chimiche di controllo; per queste ragioni è quasi sempre difficile identificare il vero momento iniziale della malattia. Questo potrà essere indirettamente ricavato dal grado di danno d'organo rilevato: se, per esempio, le arterie retiniche (le quali subiscono danni specifici dall'iperglicemia cronica) a un esame del fondo dell'occhio appaiono indenni, è verosimile che l'esordio della malattia sia recente. Al contrario, indipendentemente dall'esordio della malattia, il paziente con diabete tipo 2 avrà un rischio di eventi cardiovascolari (infarto e ictus) da due a quattro volte maggiore di un soggetto non diabetico, perché le alterazioni metaboliche che lentamente lo hanno condotto al diabete hanno contribuito anche, in vario grado, alla progressione dell'aterosclerosi.
Complicanze
Le complicanze del diabete si dividono in acute e croniche. Quelle acute sono rappresentate dalla chetoacidosi diabetica e dalla sindrome iperosmolare non chetotica, entrambe espressioni di grave iperglicemia rispettivamente in pazienti insulino-privi o in pazienti con deficit insulinico relativo. Anche l'ipoglicemia (abbassamento delle concentrazioni plasmatiche di glucosio) rappresenta un tipico evento acuto, potenzialmente pericoloso, del paziente diabetico; esso tuttavia è di fatto un effetto collaterale dei farmaci usati nel diabete, definiti appunto 'ipoglicemizzanti'.
Chetoacidosi diabetica. - Può rappresentare la manifestazione iniziale del DT1, oppure essere la conseguenza di terapia insulinica non praticata, o infine può essere indotta da gravi malattie intercorrenti (infezioni, infarto, ictus, embolia polmonare). È caratterizzata da grave disidratazione, acidosi metabolica (patologica riduzione del pH plasmatico) e disordini degli elettroliti plasmatici per perdita di sodio, potassio e fosfati. La disidratazione è legata alla già descritta poliuria per aumento della diuresi osmotica: in condizioni normali il glucosio viene filtrato dal glomerulo renale e riassorbito a livello tubulare, cosicché non si ha glucosio nelle urine. Quando la glicemia supera i 180 mg/dl, vengono saturate le capacità di riassorbimento tubulare del glucosio e una parte prosegue il suo passaggio nel rene fino a essere emesso nelle urine (glicosuria); poiché il glucosio è osmoticamente attivo, esso 'trattiene' una parte dell'acqua che altrimenti verrebbe riassorbita aumentando la perdita di liquidi per via urinaria. Per quanto riguarda l'acidosi, essa consegue alla grave carenza insulinica che induce la mobilizzazione dei grassi dal tessuto adiposo, i quali a livello epatico, per complesse alterazioni metaboliche, alimentano la cosiddetta 'chetogenesi', cioè la sintesi massiva di corpi chetonici (acido β-idrossibutirrico, acido aceto-acetico e acetone), che riversati nel circolo ematico sovrastano le capacità dei sistemi tampone inducendo grave acidosi. La perdita degli elettroliti consegue all'aumento della diuresi; in particolare il potassio, il cui ingresso all'interno delle cellule è stimolato dall'insulina, tende a essere confinato nello spazio extracellulare e a essere eliminato con le urine. Il complesso delle alterazioni descritte causa grave astenia, crampi muscolari, ipotensione fino allo shock, annebbiamento della vista, dolori addominali, nausea e vomito, progressiva alterazione dello stato di coscienza (con riduzione del senso della sete e mancata reintroduzione dell'acqua persa con le urine) fino al coma.
Sindrome iperosmolare non chetotica. - Il quadro fisiopatologico e clinico è in parte sovrapponibile a quello della chetoacidosi, con l'importante differenza dell'assenza dell'iperchetonemia, che riesce a essere inibita da una residua secrezione insulinica. Il quadro è quindi dominato da grave disidratazione e iperglicemia, che determinano un aumento patologico dell'osmolarità plasmatica. Anche in questo caso, la mancata pronta correzione terapeutica delle suddette alterazioni (con insulina, liquidi ed elettroliti per via endovenosa) può condurre al coma e all'exitus.
Le complicanze croniche si dividono in microangiopatiche, neuropatiche e macroangiopatiche.
Complicanze microangiopatiche. - Si sviluppano nei piccoli vasi sanguigni, come arteriole e capillari, che presentano alterazioni sia strutturali (ispessimento della membrana basale, esile trama di sostegno che avvolge le cellule endoteliali, che rappresentano lo strato più interno e a diretto contatto con il sangue), sia funzionali (aumento della permeabilità capillare con stravaso delle proteine plasmatiche). I microvasi più colpiti sono quelli della retina (retinopatia), del glomerulo renale (nefropatia) e dei nervi (cosiddetti vasa nervorum) che contribuiscono allo sviluppo della neuropatia diabetica. Il danno è la conseguenza diretta del cronico aumento dei livelli glicemici ed è direttamente correlato al grado e alla durata dell'iperglicemia media. Dal punto di vista patogenetico il danno è causato dall'aumentato flusso di glucosio all'interno di quelle cellule (tra cui, appunto, quelle endoteliali), nelle quali esso entra liberamente senza il controllo dell'insulina; in tali cellule il conseguente aumento del metabolismo ossidativo è associato alla generazione di radicali liberi dell'ossigeno, che quando superano il potere di neutralizzazione da parte dei sistemi antiossidanti aumentano il cosiddetto 'stress ossidativo' che danneggia le macromolecole biologiche quali il DNA. Attraverso complessi meccanismi molecolari, la riparazione del DNA condiziona l'attivazione di vie metaboliche anomale del glucosio (via dei polioli, glicosilazione non enzimatica, via delle esosammine, iperstimolazione della proteina-chinasi-C) che conducono all'aumentata produzione di molecole lesive sia delle cellule che le producono sia dei tessuti immediatamente circostanti.
La 'retinopatia' è la prima causa di cecità nei pazienti diabetici. Viene suddivisa in 'non proliferante' (o background) e 'proliferante'. La prima è caratterizzata dalla presenza di microaneurismi (dilatazioni sacciformi dei capillari), occlusioni e dilatazioni capillari, microemorragie e stravasi (essudati) che assumono un aspetto cotonoso. La forma proliferante è così detta perché la progressiva occlusione dei vasi retinici stimola la proliferazione di nuovi vasi che vanno incontro a rottura ‒ con conseguenti emorragie preretiniche e vitreali ‒ e a formazione di membrane fibrovascolari a significato cicatriziale, che esercitano un'abnorme trazione sulla retina con conseguente distacco. I pazienti diabetici pertanto devono sottoporsi annualmente a un esame del fondo dell'occhio.

La 'nefropatia' è una complicanza che non tutti i diabetici sviluppano (l'incidenza è di ca. il 30-35%), e ciò suggerisce l'intervento di una suscettibilità genetica. Le alterazioni descritte interessano i capillari dei glomeruli renali (le strutture vascolari deputate alla filtrazione del sangue e alla produzione di urina, fig. 4A) . Il marcatore più sensibile di nefropatia è costituito dalla presenza nelle urine di piccolissime quantità di albumina (principale proteina circolante) definita 'microalbuminuria'. Nella storia naturale della malattia la microalbuminuria si trasforma in macroalbuminuria, il glomerulo va incontro a sclerosi (glomerulosclerosi, fig. 4B) con progressiva riduzione della filtrazione e sviluppo di insufficienza renale cronica, che nei gradi avanzati richiede il trattamento dialitico in attesa di trapianto renale. I pazienti diabetici devono sottoporsi annualmente al dosaggio della microalbuminuria e della creatininemia (marcatore della funzione renale).
Complicanze neuropatiche. - Gli stessi meccanismi che intervengono nel danneggiamento del microcircolo contribuiscono allo sviluppo del danno dei nervi periferici, che quindi è anch'esso specificamente legato al grado e alla durata dell'iperglicemia; oltre agli effetti diretti sulle cellule neuronali, le lesioni neuropatiche sono anche conseguenti al ridotto apporto ematico causato dalla microangiopatia che interessa i vasi che irrorano i nervi. Il danno si manifesta con la perdita delle terminazioni nervose più fini, con la demielinizzazione (perdita della guaina di mielina che ricopre i nervi) e con la riduzione della velocità di conduzione degli impulsi. Il quadro clinico è caratterizzato, nella maggior parte dei casi, dalla cosiddetta 'polineuropatia simmetrica', che si manifesta con parestesie (sensazione di formicolii o punture), dolore, perdita della sensibilità termica e dolorifica che interessa inizialmente le estremità e che, a livello dei piedi, predispone alla formazione di lesioni cutanee con sviluppo di ulcere. L'interessamento dei nervi del sistema nervoso autonomo (che controlla le azioni involontarie dei vari organi) può indurre una serie di sintomi e segni, quali ipotensione ortostatica (abbassamento della pressione arteriosa alzandosi in piedi), gastroparesi con vomito, alterata motilità intestinale con stitichezza o diarrea, vescica atonica e non in grado di svuotarsi correttamente con infezioni urinarie ricorrenti, e disfunzione erettile (incapacità di raggiungere e/o di mantenere un'erezione sufficientemente prolungata per consentire un rapporto sessuale).
Complicanze macroangiopatiche. - Al contrario di quelle microangiopatiche le complicanze macroangiopatiche o macrovascolari, interessanti le arterie di grande e medio calibro, non sono specifiche del diabete ma si identificano nella malattia aterosclerotica, che si sviluppa anche in presenza di altri fattori di rischio quali obesità, alterazioni della colesterolemia caratterizzate da alto colesterolo-LDL e basso colesterolo-HDL, ipertrigliceridemia, ipertensione arteriosa, tabagismo e sedentarietà. Per tali motivi il DT2, che di fatto nella grandissima maggioranza dei casi si associa a molte di tali alterazioni, è gravato, rispetto al DT1, dallo sviluppo precoce e più aggressivo di aterosclerosi e quindi a un maggior rischio di andare incontro a eventi cardio- e cerebro-vascolari. L'aterosclerosi è causata dal progressivo accumulo di lipidi nella parete delle arterie, definita 'placca ateromasica', con progressivo restringimento del calibro delle arterie e conseguente riduzione del flusso di sangue, che si manifesta clinicamente con l'angina pectoris, se sono interessate le coronarie, con sintomi da insufficienza cerebrovascolare, se sono interessate le carotidi, e infine con claudicatio intermittens (o zoppia intermittente, cioè manifestazione dolorosa, solitamente localizzata al polpaccio, che compare dopo aver camminato per un numero di metri relativamente costante), se sono interessate le arterie che irrorano gli arti inferiori. Con l'aumentare delle dimensioni della placca aumenta anche il rischio di rottura della stessa che, se avviene nelle coronarie o nelle carotidi, determina rispettivamente infarto e ictus, mentre a livello delle gambe può essere responsabile di gangrena con rischio di amputazione.
Terapia
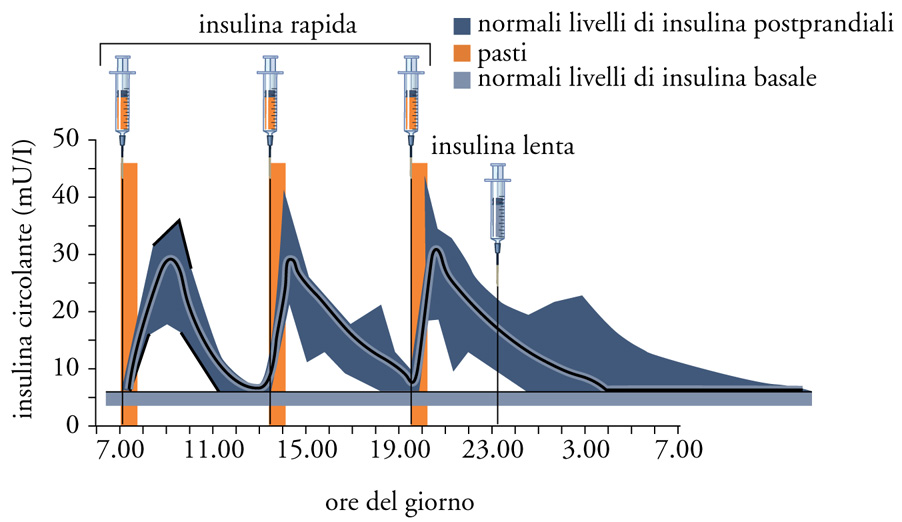
Terapia del diabete tipo 1. - Si identifica con la terapia insulinica, che viene praticata con multiple iniezioni sottocutanee (una di insulina rapida per ognuno dei tre pasti principali, e una di insulina lenta la sera prima di coricarsi); attualmente essa si giova di insuline modificate (grazie alle tecniche di DNA ricombinante) che vengono incontro alla duplice e opposta esigenza da un lato di essere rapidamente assorbite per far fronte ai picchi glicemici postprandiali e, dall'altro, di consentire un lento e costante assorbimento durante le 24 ore al fine di garantire un'adeguata insulinizzazione basale (fig. 5). Per prevenire e/o ritardare la comparsa delle complicanze del diabete, specie nei soggetti più giovani, è indicato il cosiddetto 'trattamento insulinico intensivo' che prevede l'aggiustamento del dosaggio sulla base di frequenti determinazioni glicemiche effettuate grazie a piccoli apparecchi portatili, che utilizzano quantità minime di sangue capillare (ottenute pungendo i polpastrelli con microaghi). L'effettivo buon controllo andrà verificato ogni tre-quattro mesi mediante il dosaggio dell'emoglobina glicosilata: il grado di glicosilazione dell'emoglobina è un indice della glicemia media dei tre mesi precedenti. Nonostante una corretta alimentazione debba fare da supporto a un'adeguata terapia insulinica, il paziente con DT1 non in sovrappeso non ha bisogno di seguire diete particolari, ma deve adeguarsi alle raccomandazioni alimentari per la popolazione generale, che prevedono che la maggior parte dell'energia (50-60%) provenga dai carboidrati (con l'eccezione degli zuccheri a rapido assorbimento contenuti nei dolci) e che venga limitata l'ingestione di grassi animali.
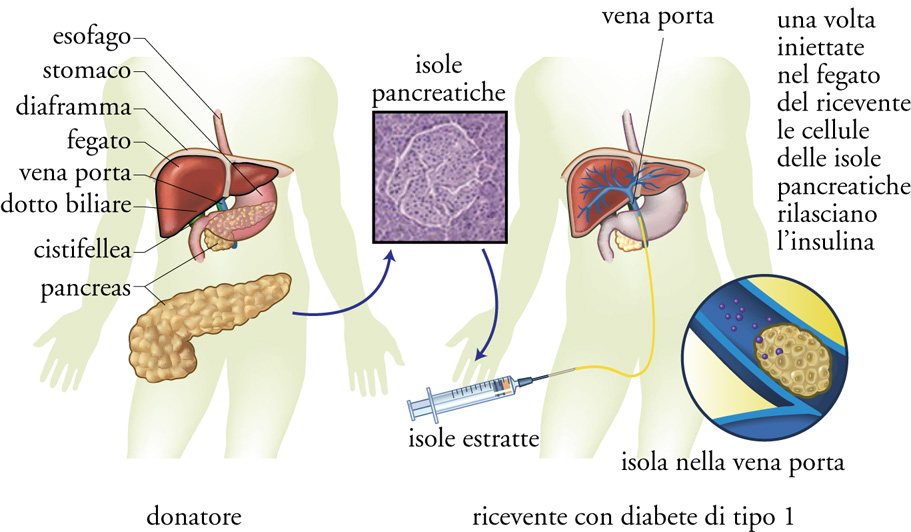
In pazienti con diabete molto instabile si può far ricorso all'infusione sottocutanea continua di insulina mediante speciali minipompe (più o meno delle dimensioni di un telefono cellulare) che, attraverso un sottile catetere di plastica impiantato sottocute nell'addome, infondono una dose continua di insulina e dosi supplementari prima dei pasti. Il trapianto di pancreas, per la necessità di seguire per tutta la vita una terapia antirigetto più dannosa del diabete stesso (qualora esso sia ben curato), è indicato solo in associazione al trapianto di rene. Tra le terapie sperimentali, il trapianto di isole pancreatiche viene riservato solo ad alcune categorie ben definite di pazienti ed effettuato in centri specializzati. Le isole pancreatiche vengono purificate dall'organo intero estratto dal donatore e infuse nella vena porta, da dove si impiantano nel fegato (fig.6). Molti sforzi si stanno facendo a livello sperimentale per tentare di modificare geneticamente le cellule β‚ affinché sia possibile evitare la pesante terapia antirigetto e affinché non venga indotta la medesima risposta immunitaria che ha determinato la malattia nel ricevente, e che di fatto vanifica gli effetti del trapianto in un periodo di tempo che raramente supera l'anno. Alla frontiera della ricerca vi sono i tentativi di utilizzare cellule staminali per indurne la differenziazione in cellule β del pancreas oppure la manipolazione genetica (terapia genica) per prevenire o curare la malattia.
Terapia del diabete tipo 2. - Differentemente dal DT1, il DT2 è caratterizzato dal progressivo e lento declino della secrezione e dell'azione insulinica causato, nella maggior parte dei casi, dall'obesità. Il cardine della terapia si basa quindi sulle modifiche dello stile di vita (dieta ed esercizio fisico) che, se attuate, possono da sole controllare la malattia. Purtroppo l'epidemia di obesità/diabete che caratterizza le società occidentali documenta l'attuale inefficacia di tali misure che, oltretutto, contribuirebbero anche al miglioramento della sindrome metabolica e quindi alla riduzione del rischio cardiovascolare. I farmaci utilizzati, definiti 'antidiabetici orali', appartengono a varie categorie qui elencate: (a) farmaci insulino-sensibilizzanti (metformina e glitazonici o tiazolidinedioni); (b) farmaci secretagoghi, che stimolano la secrezione insulinica (sulfoniluree, glinidi e analoghi dell'ormone GLP-1, Glucagon like peptide-1); (c) inibitori delle glucosidasi intestinali, che inibiscono o rallentano l'assorbimento degli zuccheri (acarbosio). Nelle fasi più avanzate della malattia è necessario associare una o più somministrazioni di insulina per far fronte al progressivo esaurimento delle cellule β. Indipendentemente dalla forma di diabete è fondamentale che il paziente venga ben educato alla gestione della malattia diabetica.
Diabete insipido
Cenni di fisiologia dell'ormone antidiuretico
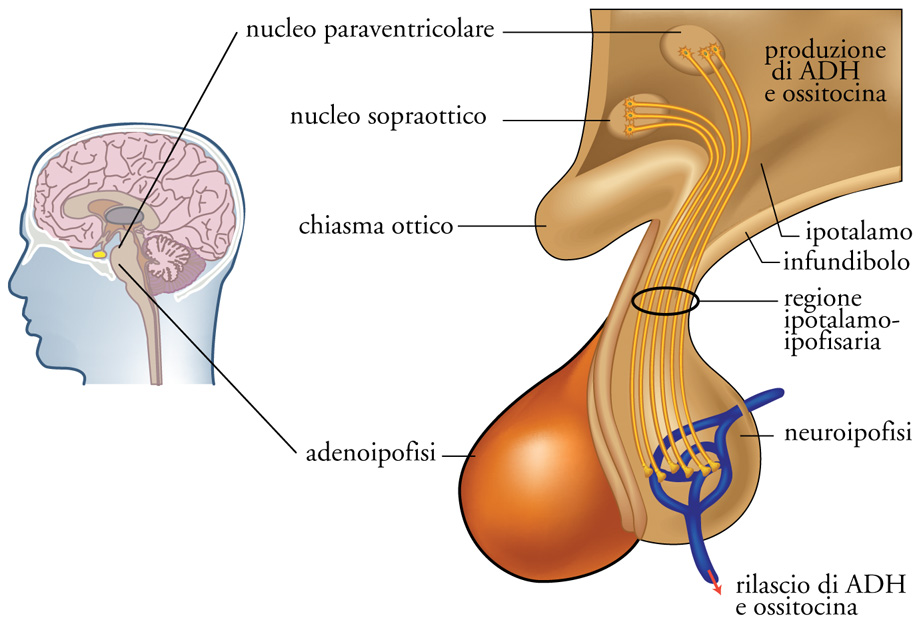
L'ormone antidiuretico interviene nel mantenimento del bilancio idrico dell'organismo e in particolare nel mantenimento dell'osmolarità (concentrazione) plasmatica entro limiti assai ristretti. In assenza di ormone antidiuretico i tubuli renali sono impermeabili all'acqua, l'urina prodotta sarà diluita e l'osmolarità plasmatica aumenterà con stimolo del senso della sete e reintegro dell'acqua persa con le urine. L'ormone antidiuretico (ADH, Antidiuretic hormone, anche definito 'vasopressina', VP, per i suoi effetti ipertensivi) viene sintetizzato da specifici neuroni ipotalamici all'interno di granuli secretori, che a loro volta vengono trasportati lungo l'assone fino alla neuroipofisi (o ipofisi posteriore) dove, in seguito a stimolazione, viene riversato nel circolo ematico (fig. 7). L'ADH agisce a livello di specifici recettori localizzati in prossimità del tubulo contorto distale del nefrone (l'unità funzionale del rene), inducendo il riassorbimento dell'acqua libera.
Classificazione del diabete insipido (DI)
Il DI si suddivide in centrale (o ipotalamico) e nefrogenico; queste due forme sono causate rispettivamente da un deficit di produzione dell'ADH e da una resistenza alla sua azione a livello renale. Entrambe le forme possono essere primitive (congenite o associate a sindromi particolari) o secondarie a traumi, tumori, malattie infiammatorie o vascolari che interessino l'ipotalamo o le regioni cerebrali contigue nel caso del DI centrale, e a malattie renali croniche, malattie metaboliche, alcune malattie sistemiche (generalizzate) e alcuni farmaci nel caso del DI nefrogenico.
Diagnosi
Il fine diagnostico mira a: (a) confermare la presenza di DI; (b) differenziare le due forme di DI (centrale, nefrogenico) dalla potomania (o polidipsia psicogena per nevrosi ossessivo-compulsive o schizofrenia); (c) identificare l'eziologia della specifica forma di DI. In presenza di una sintomatologia caratterizzata da poliuria, nicturia (enuresi nei bambini) e/o sete persistente, si impone la misurazione sia del volume urinario delle 24 ore che dell'osmolarità urinaria: se i valori confermano una poliuria ipo-osmolare (>50 ml/kg di peso corporeo nelle 24 ore con osmolarità ⟨300 mosm/l) e, contestualmente, vengono escluse altre cause di poliuria (iperglicemia, ipopotassiemia, ipercalcemia, insufficienza renale), si impone la diagnosi differenziale tra DI e polidipsia psicogena. A tal fine si effettua il 'test dell'assetamento', che di fatto saggia la capacità di concentrare le urine in corso di stress osmotico da privazione di liquidi. Qualora si ponga diagnosi di DI centrale, la diagnostica per immagini (per es., la risonanza magnetica) della regione ipotalamo-ipofisaria può individuare lesioni organiche responsabili delle forme acquisite.
Terapia
Nelle forme centrali il trattamento di scelta è rappresentato dalla desmopressina (un analogo dell'ADH privo dell'azione vasopressiva e a più lunga durata d'azione), disponibile in spray nasale, iniezioni parenterali (endovena, intramuscolare e sottocute) e compresse orali. Il DI nefrogenico non è, per definizione, responsivo alla desmopressina; vengono utilizzati i diuretici tiazidici (riducono la quantità di acqua libera nel tubulo contorto distale), i farmaci antinfiammatori non steroidei (i cosidetti FANS) o diete iposodiche. Il razionale di questi approcci risiede nella riduzione della sodiemia e della volemia, con conseguente riduzione del filtrato glomerulare e del carico di soluti nel tubulo prossimale e con aumento del riassorbimento dell'acqua in questa sede.
Bibliografia
American Diabetes Association 2006: Clinical practice recommendations, "Diabetes care", 29 (suppl. 1), 2006.
Aron 2004: Aron, David C. - Findling, James W. - Tyrrel, J. Black, Hypothalamus and pituitary gland, in: Basic and clinical endocrinology, 7. ed., edited by Francis S. Greenspan and David G. Gardner, New York-London, McGraw-Hill, 2004.
Di Mario 2000a: Diabetes in the new millennium, edited by Umberto Di Mario e altri, Chichester-New York, Wiley, 2000.
Di Mario 2000b: Di Mario, Umberto - Baroni, Marco G. - Sbraccia, Paolo, Patogenesi e fisiopatologia del diabete tipo 2, in: Manuale medico di endocrinologia e metabolismo, a cura di Mario Andreoli e altri, Roma, Il pensiero scientifico, 2000.
Einhorn, Rosenstock 2005: Type 2 diabetes and cardiovascular disease, edited by Daniel Einhorn and Julio Rosenstock, Philadelphia, Saunders, 2005.
Gottlieb 2004: Type 1 diabetes, edited by Peter A. Gottlieb, Philadelphia-London, Saunders, 2004.
Pickup, Williams 2003: Textbook of diabetes, edited by John C. Pickup and Gareth Williams, Malden (Mass.)- Oxford, Blackwell Science, 2003, 2 v.