
Superfici
Superfici
Fisica delle superfici di Gianfranco Chiarotti
SOMMARIO: 1. Introduzione. 2. La struttura elettronica della superficie ideale. 3. La superficie reale. 4. Tecniche sperimentali di fisica delle superfici: a) diffrazione di elettroni lenti (Low Energy Electron Diffraction: LEED); b) tecniche spettroscopiche; c) diffusione superficiale di molecole e ioni; d) tecniche di microscopia. 5. Modelli di superfici. □ Bibliografia.
1. Introduzione
È noto che i solidi (ad eccezione dei vetri e dei materiali amorfi) possiedono una struttura periodica in cui gli atomi sono disposti ai vertici di un reticolo cristallino (v. solidi, fisica dei). A causa della struttura irregolare o difettuale dei piani superficiali e anche per la diversa disposizione degli atomi di superficie, che non rispettano la simmetria dell'interno del solido, la struttura della superficie non coincide con quella di uno dei piani del reticolo del cristallo. Inoltre, a causa della diretta interazione con l'ambiente e della segregazione superficiale di impurezze, la composizione chimica della superficie è spesso diversa da quella del resto del solido.
Discende da ciò che alcune proprietà dei solidi sono in larga misura proprietà delle rispettive superfici: ad esempio l'emissione fotoelettrica, l'effetto termoionico, il chemisorbimento e le proprietà catalitiche, l'adesione, la resistenza alla corrosione, il trasporto elettronico nel canale superficiale dei dispositivi MOS (Metal Oxide Semiconductors) e, in minor misura, nei transistori, la durezza di alcuni materiali, ecc.
La fisica (e la chimica) delle superfici ha acquistato recentemente un'importanza rilevante anche dal punto di vista tecnologico. Essa ha d'altra parte ottenuto un grande impulso dallo sviluppo delle tecniche che permettono il controllo e la caratterizzazione delle superfici: la tecnica degli ultra-alti vuoti (~ 10-10 torr), la fotoemissione con radiazione di sincrotrone (v. Feuerbacher e altri, 1978), le varie spettroscopie elettroniche (spettroscopia Auger, perdita di energia degli elettroni, fotoemissione inversa, ecc.) e ottiche (v. Ibach, 1977), la diffrazione di elettroni lenti (v. Heinz e Muller, 1982) e di fasci molecolari (v. Engel e Rieder, 1982), l'epitassia con fasci molecolari (v. Voorhoeve, 1976), la microscopia a effetto tunnel (v. Carnevali e Selloni, 1984).
Nel seguito verrà discussa in dettaglio la struttura elettronica delle superfici, mettendone in evidenza l'influenza (spesso determinante) sulle proprietà superficie-dipendenti menzionate precedentemente.
Nel cap. 2 vengono richiamati alcuni concetti e risultati di fisica dei solidi, necessari, se non altro, per introdurre il linguaggio usato nella fisica delle superfici e per permettere la comprensione del problema fisico di un solido limitato (superficie ideale).
Nel cap. 3 sono descritte le superfici reali.
I principali metodi di indagine sperimentale usati nello studio delle superfici sono trattati nel cap. 4, mentre nel cap. 5 viene discusso in dettaglio il modello, su scala atomica, di una superficie di particolare interesse teorico e viene illustrato il procedimento di derivazione della struttura microscopica dai risultati sperimentali. Per una trattazione termodinamica dei processi di superficie si rimanda all'articolo superfici: Chimica delle, suppl.
2. La struttura elettronica della superficie ideale.
Da un punto di vista generale possiamo affermare che la maggior perturbazione introdotta in un solido dall'esistenza di una superficie è la rottura della simmetria traslazionale del reticolo cristallino. Anche per un'ipotetica superficie ideale, con gli atomi superficiali disposti regolarmente su un piano cristallografico con la stessa simmetria presente all'interno del solido, la mancanza di simmetria traslazionale nella direzione perpendicolare alla superficie introduce delle modificazioni della struttura elettronica che si rivelano particolarmente importanti, specie nel caso dei semiconduttori.
In un solido ideale (infinito) si può pensare che, a causa della simmetria traslazionale del reticolo, gli elettroni si muovano nel campo periodico prodotto dagli altri elettroni e dai nuclei disposti regolarmente nei vertici del reticolo cristallino. Questa approssimazione, che trascura in parte l'interazione elettrone-elettrone, mediandola con un potenziale periodico, viene chiamata teoria delle bande e dà origine, per i livelli elettronici dei solidi, a risultati particolarmente semplici, che si possono riassumere nel modo seguente.
1. Secondo la meccanica quantistica un elettrone si muove in un potenziale periodico come un'onda di vettore d'onda k (lunghezza d'onda λ = 2π/k) ed energia E = ℏω, con ℏ = h/2π = 1,054 × 10-34 J s, essendo h la costante di Planck e ω la frequenza angolare. La funzione d'onda (soluzione dell'equazione di Schrödinger) è data da
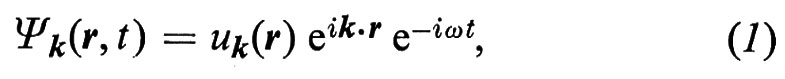
dove abbiamo indicato con r il vettore posizione, con t il tempo, con i l'unità immaginaria e con uk(r) una funzione periodica con la stessa periodicità del reticolo. La (1) rappresenta un'onda (onda di Bloch) che si propaga nella direzione di k con ampiezza che varia periodicamente. La densità di carica dell'elettrone, proporzionale a ∣ Ψk ∣2 = ∣ uk ∣2, è distribuita periodicamente, ma non necessariamente centrata sui singoli atomi. Gli elettroni, considerati come particelle localizzate nello spazio a un dato istante, possono essere ottenuti costruendo pacchetti d'onda, ossia sovrapponendo soluzioni del tipo (1) con valori di k leggermente diversi.
2. A causa della struttura discontinua e periodica del solido, non tutti i vettori d'onda k rappresentano stati (onde) fisicamente distinti. Il problema è analogo a quello della vibrazione di una catena di sferette collegate da molle. Si vede dalla fig. 1 che la vibrazione può essere descritta in modo equivalente dalla propagazione di un'onda di lunghezza d'onda 2a (‛vettore d'onda' π/a), oppure da onde di lunghezza d'onda 2a/3, 2a/5, ecc. (‛vettori d'onda' 3π/a, 5π/a, ecc., rispettivamente). Si osservi che i ‛vettori d'onda' delle varie onde equivalenti sono dati da k = π/a + 2nπ/a, n essendo un numero intero qualsiasi.
L'esempio precedentemente mostrato per un reticolo unidimensionale e per il caso della lunghezza d'onda 2a (dove a è il passo reticolare della catena di atomi) può essere immediatamente esteso a un'onda di vettore d'onda k = 2π/λ qualsiasi, osservando che, qualora si sostituisca (k + 2nπ/a) a k nella formula (1), riscritta per il caso unidimensionale, indicando con z la coordinata di posizione, si ottiene un'autofunzione che è indistinguibile dalla precedente, perchè il fattore esponenziale con esponente i2πnz/a è periodico con la periodicità del reticolo e può essere quindi conglobato nella funzione uk(z). È pertanto lecito affermare che due onde con valori di k che differiscono per un multiplo intero di 2π/a sono a tutti gli effetti indistinguibili, o, in altri termini, che k è definito a meno di n2π/a.
Il discorso precedente può essere esteso al caso di un'onda che si propaga in un cristallo la cui periodicità è caratterizzata da un reticolo di Bravais tridimensionale (v. solidi, fisica dei). Il vettore k della funzione d'onda (1) è allora definito a meno di un vettore, g, del reticolo reciproco. Tale reticolo è caratterizzato dal fatto che il prodotto scalare di uno qualsiasi dei suoi vettori g per un qualsiasi vettore di traslazione r del reticolo di Bravais è un multiplo intero di 2π (v. solidi, fisica dei).
In conseguenza di questa proprietà, è possibile definire un volume finito dello spazio reciproco, nel quale il vettore k è univocamente definito. Questa porzione di spazio, chiamata ‛prima zona di Brillouin', o più semplicemente ‛zona di Brilbuin' (BZ), si ottiene quale luogo dei punti interni a un poliedro le cui superfici di contorno tagliano perpendicolarmente a metà i vettori del reticolo reciproco che partono dall'origine. Nel caso unidimensionale la zona di Brilbuin (BZ) si riduce a un segmento compreso tra −π/a e +π/a. Nel caso bidimensionale è un poligono opportuno; nella fig. 2 si mostra a titolo di esempio il reticolo a struttura esagonale (A) e il corrispondente reticolo reciproco (B). Nel caso tridimensionale si ottengono vari tipi di poliedri, uno per ciascuno dei 14 reticoli traslazionali di Bravais (v. solidi, fisica dei).
Si può dimostrare che la BZ contiene N valori distinti di k, N essendo il numero di celle elementari del cristallo.
3. In un solido illimitato il vettore d'onda k deve essere un vettore reale (ossia con componenti tutte reali). Se una delle componenti (per es. kz) fosse immaginaria (o complessa), la funzione d'onda (1) divergerebbe esponenzialmente per z → + ∞ o per z → − ∞ a seconda del segno di kz e quindi non sarebbe normalizzabile all'unità, come richiesto dalla meccanica quantistica (v. quanti, teoria dei); l'onda d'altra parte non si propagherebbe affatto nel cristallo, ma sarebbe un'onda evanescente. Le soluzioni (1) con k immaginario vengono perciò scartate. Essendo l'energia una funzione di k, ne risulta che esistono degli intervalli permessi di energia (bande permesse), cui corrispondono valori reali di k e onde che si propagano nel cristallo, separati da intervalli proibiti (bande proibite o gaps), cui corrispondono valori immaginari di k e onde evanescenti.
4. Gli elettroni di un solido occupano, alla temperatura T = 0 K, le bande permesse di minor energia, compatibilmente con il principio di esclusione di Pauli. Ogni banda permessa contiene N stati (corrispondenti ai valori distinti di k nella BZ) e può accogliere al più 2N elettroni (con i due valori permessi dello spin). Se il solido è costituito da atomi con un numero dispari di elettroni (ad es. Na, Cu, Al, ecc.), l'ultima banda occupata sarà piena solo per metà. Il solido sarà cioè un metallo, in quanto gli elettroni trovano stati vuoti (a energia infinitamente prossima a quella posseduta) sui quali possono essere ‛eccitati' dal campo elettrico, contribuendo alla conduzione elettrica. Quando il solido è costituito da atomi con un numero pari di elettroni (ad es. C, Si, Ge, ecc.), la banda permessa più alta sarà occupata completamente e il solido sarà un isolante (o un semiconduttore). Fanno eccezione i solidi nei quali si verifica una sovrapposizione di bande permesse in alcune regioni della BZ. Ciò elimina di fatto le gaps rendendo il solido conduttore. È questo il caso, ad esempio, del Be o del Mg, che, pur avendo un numero pari di elettroni per atomo, sono dei metalli.
A temperature diverse da 0 K, alcuni elettroni vengono eccitati sui livelli vuoti, in accordo con la legge di distribuzione statistica di Fermi-Dirac. A 0 K tale distribuzione è caratterizzata da una funzione a gradino: tutti i livelli a energia minore di EF sono pieni, mentre quelli a energia maggiore sono vuoti. Per temperature T > 0 K, la distribuzione a gradino si smussa, permettendo che anche i livelli sopra EF siano parzialmente occupati. EF viene chiamata energia di Fermi e gioca un ruolo importante nella fisica delle superfici.
L'energia di un elettrone a riposo fuori dal solido viene spesso indicata con Evac. La minima energia necessaria per estrarre un elettrone dal solido è (a T = 0 K) Evac − EF e viene chiamata ‛lavoro di estrazione' o, più semplicemente, ‛funzione lavoro'.
La situazione descritta al punto 3, che caratterizza la struttura elettronica dei solidi, non è più valida per un solido limitato, ossia in presenza di una superficie. La superficie, infatti, interrompendo la simmetria traslazionale del solido, fa cadere la condizione che le energie permesse siano associate unicamente a valori reali di k. Ciò è illustrato nella fig. 3: nella fig. 3A è rappresentata schematicamente l'energia potenziale di un elettrone in un solido unidimensionale con superficie a z = 0. Evac rappresenta l'energia potenziale dell'elettrone fuori dal solido, mentre E è la sua energia totale (cinetica più potenziale), ovviamente costante. Essendo E minore di Evac, l'elettrone è confinato all'interno del solido. Infatti, secondo la meccanica quantistica, la funzione d'onda decade esponenzialmente per z 〈 0 e diventa nulla a distanza infinita.
Gli stati della (1) con k reale corrispondono a elettroni che si riflettono sulla superficie e la cui energia è (praticamente) la stessa che avrebbero nel cristallo infinito. La loro funzione d'onda è quella di un'onda di Bloch per z 〈 0 e decade esponenzialmente per z > 0.
Nel caso di k immaginario la (1) si riduce, per z 〈 0 e omettendo la dipendenza temporale, a
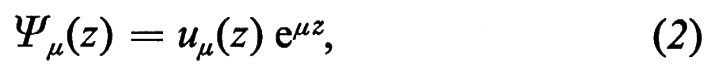
con μ = ik reale, mentre rimane un esponenziale decrescente per z > 0. Tale funzione d'onda è rappresentata schematicamente nella fig. 3B per il caso in cui sia μ > 0 (l'unico di interesse fisico). Si vede che lo stato con k immaginario rappresenta un elettrone localizzato in prossimità della superficie con energia corrispondente (proprio perché k è immaginario) alle bande proibite del solido infinito (v. Forstmann, 1971). La presenza della superficie introduce, cioè, stati nelle gaps e pertanto modifica drasticamente le proprietà dei solidi (specie degli isolanti e dei semiconduttori). Tali stati, che da un punto di vista chimico sarebbero associati ai legami non saturati degli atomi di superficie (dangling bonds), possono catturare o cedere elettroni, causando la formazione di una carica nel piano di superficie. Essa a sua volta determina un campo elettrico macroscopico schermato verso l'interno dagli altri elettroni del solido. Si ha cioè un incurvamento superficiale delle bande di energia degli elettroni, che modifica il lavoro di estrazione e il trasferimento di carica tra il solido e le molecole dell'ambiente esterno (v. Many, 1975).
3. La superficie reale.
Le considerazioni svolte nel capitolo precedente consentono di definire stati elettronici localizzati alla superficie solo nel caso di un cristallo unidimensionale e nell'ambito di un modello semplificato. Nel caso di un solido reale è necessario tener conto di numerose altre condizioni che verranno illustrate in questo capitolo.
1. Nel caso ideale tridimensionale la periodicità è mantenuta nel piano di superficie xy. Il vettore d'onda k ha componenti x, y reali e una componente z immaginaria. Gli stati di superficie sono rappresentati da onde viaggianti nel piano xy ed evanescenti nella direzione z. Essi sono definiti in una BZ superficiale bidimensionale. A causa dell'energia (cinetica e potenziale) associata al moto nel piano xy, gli stati di superficie possono avere energie non necessariamente corrispondenti a quelle delle gaps. Quando la degenerazione con gli stati dell'interno del cristallo è limitata all'energia (e non a k), si hanno veri e propri stati di superficie anche a energie diverse da quelle delle gaps.
2. A causa dell'esposizione all'ambiente esterno e della presenza di legami insaturi, caratteristica della troncatura del cristallo, la composizione chimica di una superficie è spesso assai diversa da quella dell'interno del solido. Atomi o molecole di specie estranee possono essere chemisorbiti o fisisorbiti in superficie o può verificarsi una precipitazione di impurezze nei primi strati del solido. In generale si parla di fisisorbimento o di chemisorbimento a seconda che le energie di legame atomo-superficie siano dell'ordine di 10-2 o 1 eV/atomo rispettivamente. Non esiste tuttavia una distinzione netta tra i due fenomeni, che possono essere indicati indistintamente col termine ‛adsorbimento'.
La comprensione della struttura delle superfici ha ricevuto un notevole impulso dallo studio delle cosiddette superfici ‛pulite', ossia prive di impurezze chimiche. Tali superfici possono essere ottenute in vari modi: a) mediante sfaldatura dei cristalli in ultra-alto vuoto (UHV, dell'ordine di 10-10 torr). Lo studio è in questo caso limitato alle superfici di sfaldatura, che sono, in generale, quelle di più stretto impacchettamento degli atomi; b) mediante bombardamento della superficie con ioni di un gas nobile (in generale argon) di qualche keV. Tale bombardamento (sputtering) erode i primi strati della superficie e con essi le impurezze eventualmente presenti. È necessario però ricuocere in UHV la superficie (processo di annealing) a una temperatura sufficientemente alta da far evaporare gli atomi di Ar che sono rimasti sepolti nella superficie. Queste superfici ‛ricotte' possono avere una struttura assai diversa da quelle ottenute per sfaldatura; c) mediante riscaldamento della superficie in UHV a temperature prossime alla temperatura di fusione. Questo processo è molto conveniente per i metalli alto-fondenti (per es. Ni, W), nei quali le impurezze chemisorbite hanno energie di legame inferiori a quelle della matrice e vengono eliminate per evaporazione. La struttura di queste superfici è simile a quella delle superfici ‛ricotte'.
In contrapposizione alle superfici ‛pulite' che abbiamo testé descritto, le altre superfici vengono chiamate ‛reali'. Esse vengono variamente trattate con soluzioni acide (etching) per eliminarne le parti più danneggiate, senza poter evitare però l'adsorbimento delle molecole della soluzione di etching.
3. La disposizione degli atomi di superficie è spesso diversa da quella dell'interno del solido (il bulk) sia per quanto riguarda le distanze interatomiche sia per quanto riguarda la simmetria traslazionale. Nei due casi si parla rispettivamente di rilassamento e di ricostruzione superficiale. La ricostruzione si può verificare anche nel caso di atomi adsorbiti, che si dispongono spesso secondo un reticolo diverso da quello del substrato e qualche volta in modo non commensurato con le distanze reticolari del substrato.
Nella maggior parte delle superfici ricostruite la cella elementare è un multiplo della cella ideale del piano superficiale. Le superfici ricostruite vengono perciò indicate facendo seguire agli indici di Miller del piano cristallografico l'espressione m × n con m, n interi positivi che indicano le dimensioni della cella ricostruita rispetto a quelle della cella ideale. Ad esempio Si (111)-2 × 1 significa che il piano (111) del silicio è ricostruito in modo che la cella elementare superficiale (costituita nel caso ideale da rombi con i lati nelle direzioni [101] e [-110] o equivalenti) ha uno dei due lati doppio di quello della cella ideale. La ricostruzione 2 × 1 dell'ipotetico cristallo bidimensionale della fig. 2A è mostrata nella fig. 2C. In essa si immagina che gli atomi indicati con cerchi vuoti siano sollevati e quelli indicati con cerchi pieni siano abbassati rispetto al piano della figura. Gli atomi ○ e ???17??? vengono così a occupare posizioni non equivalenti. Esistono ovviamente molti modi di rendere non equivalenti gli atomi ○ e ???17???, cosicché il problema della ricostruzione superficiale è in generale molto complesso e di non facile soluzione. Nella figura 2D è mostrato il reticolo reciproco della superficie ricostruita 2 × 1. La disposizione dei siti reticolari nelle figg. 2A e 2B è quella che si riscontra nella superficie (111) non ricostruita del Si. La fig. 2C si riferisce invece a una ipotetica ricostruzione 2 × 1 che non si verifica nella realtà; essa è indicata solo a titolo di esempio per la sua semplicità (per maggiori dettagli v. cap. 5).
Qualora le celle elementari della superficie ricostruita risultino ruotate, si aggiunge l'angolo di rotazione; in questo caso però i numeri m ed n non sono necessariamente interi. Nella fig. 4 è rappresentata una ipotetica superficie ricoperta da un monostrato di atomi (o molecole) adsorbiti. La cella elementare della struttura degli atomi adsorbiti (a sinistra nel disegno) può essere indicata come √-2 × √-2 − 45°. È possibile però individuare una cella non elementare che ripetuta periodicamente riproduce la stessa struttura; essa è mostrata sulla destra della figura e può essere indicata con c(2 × 2), dove la lettera c sta a indicare che la cella è centrata rispetto alla cella del substrato.
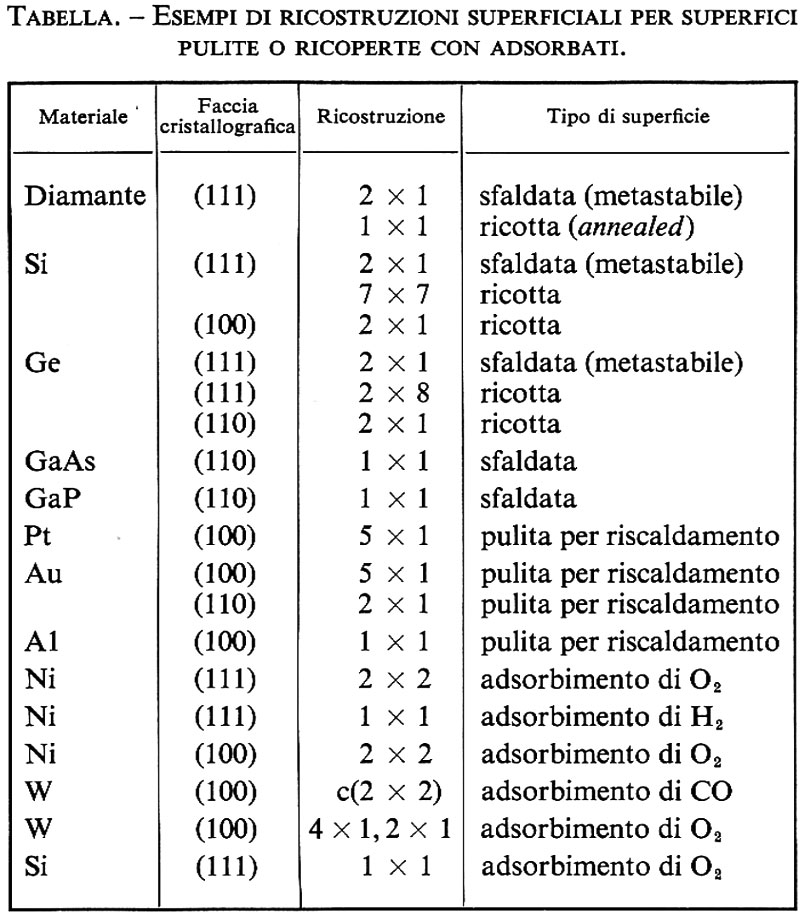
Nella tabella sono riportate alcune ricostruzioni superficiali per superfici pulite o ricoperte con adsorbati (v. Tosatti, 1976; v. Pendry, 1974).
La ricostruzione è causata dalla diversa condizione di minimo dell'energia libera che si ha in superficie a causa dell'assenza di parte delle interazioni tra gli atomi. Infatti, se la ricostruzione avviene nel vuoto (o in condizioni di debole pressione), l'equilibrio termodinamico si verifica quando l'energia libera U − TS è minima, U, S e T essendo rispettivamente l'energia interna, l'entropia e la temperatura termodinamica del sistema. Poiché le energie elettroniche sono la parte preponderante dell'energia del sistema, la conoscenza dettagliata degli stati di superficie è indispensabile qualora si voglia prevedere teoricamente la ricostruzione superficiale.
A causa del termine entropico nell'energia libera, le superfici ricostruite possono subire delle vere e proprie transizioni di fase al variare della temperatura. Qualora esistano più minimi dell'energia libera, si possono osservare delle superfici metastabili. Per esempio, un cristallo di silicio sfaldato a temperatura ambiente, in modo da esporre il piano (111), presenta una ricostruzione del tipo 2 × 1 con la cella elementare raddoppiata in una delle tre direzioni equivalenti [112]. Tale superficie è metastabile e si trasforma in una superficie stabile, ricostruita 7 × 7, mediante riscaldamento (annealing) a temperature superiori a 350 °C. La superficie 7 × 7 presenta dei blocchi di 49 atomi che si ripetono periodicamente; la sua cella elementare è cioè del tipo n × n, con n = 7.
4. Indipendentemente dalla ricostruzione e dalla contaminazione, le superfici dei solidi presentano dei difetti su scala semimicroscopica (gradini, isole, pozzi, terrazze, pieghe, ecc.), dovuti all'impossibilità di rappresentare un'entità fisica come la superficie con un piano geometrico. Essi sono illustrati schematicamente nella fig. 5 e, per la superficie (111) del silicio, nella fig. 6. I vari spigoli che sono associati ai difetti rappresentati nelle figg. 5 e 6 determinano una struttura elettronica locale diversa da quella della superficie non difettosa, a causa del numero diverso di dangling bonds, come si vede chiaramente dalla struttura riportata nella fig. 6 in alto.
La struttura difettuale di una superficie dipende in genere dal modo in cui essa viene creata e dall'energia disponibile in tale processo. In termini generali si può dire che la superficie tende a esporre, per quanto possibile, i piani di grande impacchettamento cui è associata una minore energia per unità di area. Ciò è evidente nell'esempio della fig. 6, in cui un certo disallineamento del piano superficiale rispetto al piano (111) di alto impacchettamento determina la comparsa di gradini (steps).
L'articolazione dei gradini in strutture contorte (kinks) è favorita termodinamicamente, in quanto aumenta l'entropia configurazionale del sistema.
La diversa struttura elettronica locale di una data superficie può avere un'importanza rilevante per l'interazione con le molecole dell'ambiente esterno, per esempio nel chemisorbimento o nel processo di crescita del cristallo.
5. Quando il contatto non avviene tra un solido e un gas (o un liquido), ma tra due solidi, si parla di interfaccia solida, una struttura che presenta alcune affinità con quella delle superfici libere.
La diversità (mismatch) dei parametri reticolari e delle strutture elettroniche dei due componenti (per esempio diversità delle gaps nelle eterogiunzioni), la carica spaziale nella regione dell'interfaccia, l'interdiffusione dei componenti, ecc. giocano un ruolo essenziale nella fisica delle interfacce (v. Margaritondo, 1983). Pur essendo questo argomento molto importante nella tecnologia dei dispositivi a semiconduttore, ci limiteremo nell'articolo a questo breve accenno, in quanto una sua discussione ci porterebbe troppo lontano, impedendoci di presentare un campo quanto più possibile omogeneo e suscettibile di una descrizione unitaria.
4. Tecniche sperimentali di fisica delle superfici.
Prima di procedere a una discussione più approfondita dei modelli di ricostruzione delle superfici, è necessario descrivere brevemente le principali tecniche correntemente usate e la loro rilevanza e sensibilità per lo studio della struttura e delle proprietà elettroniche superficiali.
Grossolanamente potremo classificare tali tecniche a seconda del tipo di sonda (probe) usata, ossia a seconda che vengano inviati contro la superficie fasci di elettroni, di fotoni, di molecole, di ioni, ecc. Queste particelle scambiano con la superficie energia e impulso e possono estrarne atomi e ioni. A seconda che venga o meno scambiata energia con la superficie, le tecniche si classificano inoltre in spettroscopiche o diffrattive. Le tecniche topografiche, infine, sono quelle tecniche che forniscono un'immagine della superficie.
Il capitolo tratterà dei seguenti argomenti: a) diffrazione degli elettroni lenti; b) tecniche spettroscopiche (spettroscopia ottica, spettroscopie elettroniche, spettroscopia di fotoemissione); c) scattering di molecole e ioni; d) tecniche di microscopia (topografiche).
Ciascuna di queste tecniche mette in luce particolari aspetti della struttura superficiale e ha una sensibilità più o meno grande nei confronti delle proprietà di superficie. In linea del tutto generale potremo dire che le tecniche che fanno uso di elettroni e di fasci molecolari (o di ioni di bassa energia) sono quelle che presentano la massima sensibilità superficiale, data la forte interazione di queste particelle con gli atomi della superficie: per esempio, la sezione d'urto di un elettrone è circa 105 volte più grande di quella di un fotone dei raggi X. Questa grande interazione produce però anche effetti indesiderati, come, ad esempio, urti multipli, evaporazione o erosione della superficie, ecc.
L'interazione di un elettrone con la superficie può essere massimizzata variandone l'energia cinetica, come si vede dalla fig. 7, in cui vengono riportati, per diversi materiali, i cammini liberi medi degli elettroni in funzione, appunto, della loro energia cinetica (v. Riviere, 1973). Si vede che, per elettroni di energia dell'ordine di 100 eV, il cammino libero medio è dell'ordine di 5 Å (1 Å = 10-8 cm), ossia dell'ordine delle distanze reticolari. Elettroni di tale energia riflessi dalla superficie senza perdita di energia (o con perdite controllate) permettono di ricavare informazioni dirette sui primi strati atomici del solido.
a) Diffrazione di elettroni lenti (Low Energy Electron Diffraction: LEED).
Come è noto, un elettrone libero, di impulso p = mv, si comporta come un'onda con una lunghezza d'onda data dalla formula di de Broglie λ = h/mv. Per un elettrone di 100 eV, la lunghezza d'onda è di circa 1,23 Å, ossia dello stesso ordine delle distanze interatomiche. Un fascio di elettroni ditale energia, riflesso da una superficie cristallina, subisce cospicui fenomeni di diffrazione: le intensità diffratte si rafforzano in certè direzioni, quando le differenze di cammino tra i fascetti diffusi dai vari atomi sono pari a un numero intero di lunghezze d'onda. Gli elettroni diffratti sono raccolti su uno schermo fluorescente simile a quello televisivo e danno origine a figure di diffrazione (v. figg. 8 e 9) simili alle figure di Lane ottenute con raggi X per i solidi tridimensionali.
La teoria della diffrazione (v. Pendry, 1974) fa intervenire il reticolo reciproco descritto nel cap. 2. Per un reticolo bidimensionale si trova che l'intensità diffratta ha dei massimi molto acuti se
k′∣ ∣ − k∣ ∣ = gmn, (3)
dove k∣ ∣ e k′∣ ∣ sono le componenti del vettore d'onda dell'elettrone incidente e di quello riflesso parallele al piano superficiale e gmn è uno dei vettori del reticolo reciproco della superficie. Essendo la diffrazione un processo elastico, si ha inoltre k2 = k′2.
Se g = 0, si ha il caso della riflessione ottica. Per un reticolo unidimensionale si ottiene immediatamente dalla (3)
a(cos θ′ − cos θ) = nλ (4)
a essendo il passo del reticolo (o distanza tra i centri diffondenti), θ e θ′ gli angoli di incidenza e di diffrazione (definiti convenzionalmente come gli angoli tra i fasci incidente e diffratto e la superficie del reticolo) e n un intero qualsiasi chiamato ‛ordine di diffrazione'. La (4) è la formula usuale per la diffrazione da un reticolo ottico, che è, di fatto, un reticolo unidimensionale.
Si vede dalla (3) che, se l'incidenza del fascio di elettroni è normale (k∣ ∣ = 0), la figura di diffrazione consiste in un insieme di punti identico a quello del reticolo reciproco. Per una superficie come quella del Si (111) si dovrebbe ottenere cioè una figura di diffrazione che riproduca il reticolo esagonale della fig. 2A. La fig. 8 mostra i risultati del LEED per il Si (111) ricotto (annealed) a temperature superiori a 350 °C. Oltre alle macchie che individuano la BZ del reticolo esagonale della superficie ideale (che sono le macchie più intense e quella centrale nascosta dall'ombra del cannone elettronico), si vedono altre macchie, che dividono il lato dell'esagono in 7 parti. L'esperienza mostra cioè che la superficie del Si (111) trattata come descritto presenta una ricostruzione 7 × 7.
La superficie del Si (111) ottenuta per sfaldatura a temperature inferiori a 350 °C presenta invece una figura di diffrazione del tipo 2 × 1, riportata nella fig. 9. Nella parte destra della figura viene schematizzata la figura di diffrazione, ossia il reticolo reciproco e la zona di Brillouin (rettangolare) ottenuta bisecando i vettori più piccoli del reticolo reciproco. Le macchie dovute alla ricostruzione (half-order spots) sono quelle più piccole.
Le figure mostrano chiaramente la potenza del metodo LEED nello studio della ricostruzione. L'analisi delle figure LEED non è però sufficiente a definire univocamente il modello di ricostruzione della superficie. Infatti esistono a priori molti modelli di ricostruzione che presentano la corretta simmetria rivelata dal LEED. Informazioni più dettagliate si ottengono con il metodo del LEED dinamico (v. Pendry, 1974), in cui viene fatta variare la lunghezza d'onda degli elettroni, cambiando il potenziale acceleratore nel cannone elettronico. La teoria prevede, per ogni macchia, una dipendenza dell'intensità I dall'energia degli elettroni, che è univocamente determinata dal valore V del potenziale acceleratore. Il fascio riflesso viene raccolto da una piccola coppa di Faraday e la corrente di elettroni è misurata opportunamente. Si ottengono delle curve simili a quella mostrata nella fig. 10 (curva superiore) per la superficie (100) dell'Al non ricostruita (v. Jona, 1970). La curva sperimentale è poi confrontata con quelle teoriche, ottenute assumendo che le distanze reticolari in superficie rimangano invariate (curva inferiore a tratto pieno), oppure che il piano di superficie subisca una contrazione del 2% (curva tratteggiata) o una espansione del 5% (curva punteggiata). Si vede che la superficie Al (100) non presenta rilassamento nei limiti dell'errore sperimentale.
Il metodo del LEED dinamico è però indiretto (essendo basato sul confronto tra teoria ed esperienza) e non sempre dà risultati consistenti, nel caso delle superfici ricostruite.
La diffrazione di elettroni lenti è anche sensibile ai difetti microscopici della superficie, che allargano o sdoppiano le macchie del LEED. Per esempio, in presenza di un'alta densità di gradini, tutti allineati in una data direzione, le intensità delle onde diffratte interferiscono, dando origine a una struttura a doppietti delle macchie LEED (v. Henzler, 1970).
b) Tecniche spettroscopiche.
Nelle tecniche spettroscopiche la ‛sonda', sia essa particella o fotone, scambia energia con il sistema, eccitandone i livelli elettronici o vibrazionali. Potremo suddividere tali tecniche in: spettroscopia ottica di superficie, spettroscopie elettroniche, spettroscopia di fotoemissione.
Spettroscopia ottica di superficie. - La luce riflessa da una superficie penetra nel solido per una distanza dell'ordine di α-1, α essendo il coefficiente di assorbimento ottico. Per un solido, a non è mai superiore a 105 ÷ 106 cm-1, per cui la penetrazione della luce è sempre maggiore di 100-1.000 Å; l'interazione della luce con la superficie è perciò, in generale, scarsa. Sono state tuttavia messe a punto delle tecniche ottiche assai sensibili che hanno dato risultati importanti per la fisica delle superfici dei semiconduttori.
Si può ottenere un notevole aumento di sensibilità ricorrendo 1) al metodo delle riflessioni totali multiple interne; 2) a metodi differenziali; 3) a misure ellissometriche.
1. Riflettività totale interna. Sagomando opportunamente il campione, è possibile far incidere il fascio di luce sulla superficie interna, con angoli di incidenza superiori all'angolo limite (v. Harrick e Beckmann, 1974). Il campione si comporta come una guida di luce e le riflessioni sulla superficie da studiare possono essere dell'ordine del centinaio, con un notevole aumento della sensibilità di superficie. Il metodo è particolarmente adatto a studiare la struttura elettronica e vibrazionale delle molecole adsorbite, in quanto, nella riflessione totale, la luce penetra nel secondo mezzo per distanze dell'ordine della lunghezza d'onda, interagendo con ciò che sta sulla superficie. La fig. 11 mostra lo spettro vibrazionale (infrarosso) di idrogeno e deuterio chemisorbiti su una superficie di Si (100)-2 × 1 (v. Chabal e Raghavachari, 1984). I due picchi in ciascuna struttura si riferiscono all'eccitazione dei modi di stretching simmetrici e antisimmetrici dei legami Si-H e Si-D.
Il metodo delle riflessioni multiple interne è stato anche utilizzato per lo studio delle superfici ‛pulite', nelle quali gli stati intrinseci superficiali svolgono il ruolo delle molecole adsorbite (v. Chiarotti e altri, 1968 e 1971).
2. Riflettività differenziale. La spettroscopia col metodo delle riflessioni interne è ovviamente limitata al campo spettrale per cui il solido è trasparente, ossia, nei semiconduttori, alle energie inferiori alla gap. Per energie superiori si può usare la tecnica della riflettività esterna differenziale. In questo caso l'aumento di sensibilità non può essere ottenuto con le riflessioni multiple (la luce non può riflettersi totalmente, essendo l'indice di rifrazione del campione maggiore di quello del vuoto). Si può invece usare un metodo differenziale, confrontando la riflettività della superficie ‛pulita' con quella di una superficie in cui gli stati di superficie siano stati distrutti saturando i dangling bonds con elettroni di atomi (o molecole) chemisorbiti (tipicamente H e O). Le tecniche elettroniche differenziali permettono di ottenere l'aumento di sensibilità richiesto (v. Chiaradia e altri, 1978).
Nella fig. 12 è mostrato lo spettro di riflettività differenziale (ΔR/R) per una superficie ‛pulita' di Si (111)-2 × 1 per due orientazioni del vettore elettrico della luce polarizzata, riferite alla struttura 2 × 1 della superficie ricostruita (v. Chiaradia e altri, 1984; v. Selci e altri, 1985). Si nota a 0,45 eV un picco molto netto dovuto a transizioni da stati superficiali pieni a stati vuoti e inoltre una grandissima anisotropia ottica della superficie.
Dal punto di vista teorico è possibile descrivere la riflettività di una superficie nell'ambito dell'elettromagnetismo classico con un modello a tre strati (ambiente esterno, superficie di spessore d, bulk) come quello mostrato nella fig. 13. A ciascuno dei tre mezzi è associata una costante dielettrica complessa εéj = ε′j + iεj′′ (j = 1, 2, 3), dove ε′′, la parte immaginaria della costante dielettrica, è responsabile dell'assorbimento dell'energia ed ε′ della dispersione. La teoria viene sviluppata (v. Mclntyre e Aspnes, 1971) imponendo la continuità delle componenti parallele del campo elettrico dell'onda alle interfacce, e calcolando l'interferenza, in una data direzione, dei raggi riflessi (v. fig. 13). L'effetto degli stati elettronici (o vibrazionali) della superficie può essere valutato dalla dipendenza spettrale di ΔR = R(d) − R(0), dove R(0) rappresenta la riflettività della superficie senza stati elettronici (o vibrazionali).
Nel caso di un isolante (o di un semiconduttore), per energie dei fotoni incidenti al di sotto della gap (per cui εé3 è reale), si ottiene, per incidenza normale e per d ≪ λ,
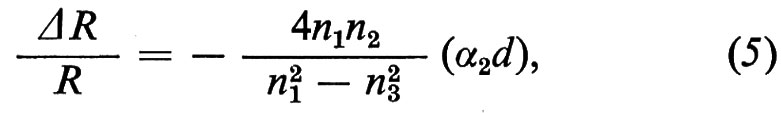
dove nj (j = 1, 2, 3) è l'indice di rifrazione del mezzo j-esimo, α2 il coefficiente d'assorbimento ottico del mezzo 2 e (α2 d) la cosiddetta ‛costante ottica di assorbimento superficiale'. Il coefficiente α è legato alla parte immaginaria della costante dielettrica dalla relazione α = (2π/nλ)ε′′.
La (5) vale sia per la riflettività esterna sia per quella interna. Passando dall'una all'altra (ossia scambiando gli indici 1 e 3), ΔR/R cambia di segno; se il mezzo esterno è il vuoto, l'effetto è inoltre più grande nella riflettività interna di un fattore pari a n3. Per fotoni di energia superiore a quella della gap le formule sono un po' più complicate.
Nel caso di raggi X la penetrazione è troppo grande per poter prevedere una sufficiente sensibilità alla superficie. Tuttavia, nel caso che la variazione dell'intensità riflessa venga rivelata misurando la corrente di elettroni fotoemessi, il piccolo cammino libero medio degli elettroni nel solido (v. fig. 7) permette di accrescere notevolmente la sensibilità del metodo. Metodi come il SEXAFS (Surface Extended X-ray Absorption Fine Structure) vengono attualmente sviluppati e potranno dare informazioni preziose specie sulle distanze reticolari in superficie (v. Citrin e altri, 1978; v. Comin e altri, 1983). Tali metodi sono affini a quelli di fotoemissione che verranno descritti in seguito.
3. Ellissometria. La luce riflessa da una superficie a incidenze non normali risulta in genere polarizzata ellitticamente. Se il mezzo riflettente assorbe in parte la radiazione, l'orientazione degli assi dell'ellisse e la sua eccentricità possono variare. I metodi ellissometrici sono molto sensibili a piccole variazioni delle costanti ottiche del campione e sono stati perciò variamente impiegati nello studio delle superfici, specie per seguirne il ricoprimento (v. Aspnes, 1976).
Spettroscopie elettroniche. - Nelle varie spettroscopie elettroniche, un fascio di elettroni monocromatici viene inviato contro una superficie e l'energia cinetica degli elettroni riflessi (o trasmessi) viene analizzata mediante opportuni dispositivi. A causa della grande interazione degli elettroni di bassa energia con gli atomi, le spettroscopie elettroniche presentano una notevole sensibilità alla superficie. Tale sensibilità può essere inoltre massimizzata scegliendo energie degli elettroni incidenti corrispondenti al minimo della curva della fig. 7.
Lo spettro di energia degli elettroni riflessi da una superficie è simile a quello della fig. 14, che si riferisce a un'energia dei primari di 1.500 eV. A energie immediatamente inferiori al picco elastico si possono osservare delle strutture associate agli elettroni che hanno perso energia interagendo con le vibrazioni reticolari e (a energie più basse) con gli stati elettronici del sistema. Cambiando l'energia degli elettroni primari queste strutture si muovono rigidamente insieme al picco elastico. Il loro studio costituisce l'oggetto della spettroscopia di perdita di energia degli elettroni o EELS (Electron Energy Loss Spectroscopy). Altre strutture si presentano invece sempre alla stessa energia (caratteristica del solido o delle molecole adsorbite) e costituiscono l'oggetto della spettroscopia Auger o AES (Auger Electron Spectroscopy). Modulando l'energia dei primari e sfruttando la diversa dipendenza dei due tipi di effetti da tale energia è possibile discriminarli con tecniche elettroniche.
Nella fig. 14 il picco a piccole energie è dovuto agli elettroni secondari che hanno perso la loro energia in urti multipli successivi. La curva non va all'infinito per energie tendenti a zero a causa del taglio operato dalla funzione lavoro (o lavoro di estrazione) del solido. L'analisi degli elettroni secondari è di grande interesse per lo studio della funzione lavoro e delle sue modificazioni durante i processi di chemisorbimento.
1. Perdite di energia degli elettroni. Questa tecnica permette in linea di principio di determinare le energie di tutte le eccitazioni elementari di un solido e della sua superficie e si confronta con la spettroscopia ottica prima descritta (v. Raether, 1965; v. Froitzheim, 1977).
Un elettrone di bassa energia che si propaga in un solido genera un campo elettrico le cui componenti di Fourier alle varie frequenze interagiscono con gli altri elettroni del solido in modo simile a un'onda elettromagnetica. La teoria mostra (v. Froitzheim e altri, 1975) che l'intensità di perdita di energia nel bulk è proporzionale a − Im(1/εé), mentre per una superficie (in cui lo schermo elettrostatico dovuto alla polarizzabilità degli atomi è minore in quanto il dielettrico occupa solo una metà dello spazio) è proporzionale a
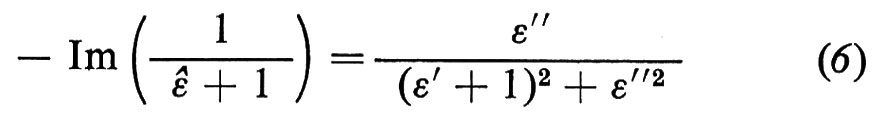
(con Im(z) si indica la parte immaginaria del numero complesso z).
Abbiamo visto, trattando la spettroscopia ottica, che ΔR/R è proporzionale a ε′′. La formula (6) mostra pertanto che le misure di perdita di energia degli elettroni sono simili alle misure ottiche; le differenze, assai piccole, discendono dalla dipendenza spettrale del denominatore, che, in generale, sposta i picchi a energie più grandi di quelle ottiche. Infatti il denominatore della (6) è minimo per ε′ = − 1, ossia a energie più grandi di quelle del picco di ε′′. Questo fatto risulta evidente dal confronto tra la fig. 15, dove è riportato lo spettro delle perdite di energia per il Si (111)-2 × 1 (v. Rowe e altri, 1975), e la fig. 12, relativa alle misure ottiche. Il picco principale associato alle transizioni dei dangling bonds si osserva a 0,52 eV nelle perdite di energia e a 0,45 eV nello spettro ottico. Anche le altre strutture comuni sono simili e dimostrano la sostanziale equivalenza dei due metodi.
2. Spettroscopia Auger. La spettroscopia Auger si fonda sull'effetto Auger (dal nome del fisico francese P. Auger), che si manifesta negli atomi quando, mediante urto elettronico o con un fotone di sufficiente energia, viene strappato un elettrone degli strati interni. Il ‛buco' è riempito da un elettrone degli strati più esterni e l'energia resa disponibile è trasferita a un altro elettrone, che, essendo espulso, contribuisce alla corrente esterna (v. Rivière, 1973; v. Carlson, 1978). Poiché nel fenomeno sono coinvolti principalmente gli elettroni interni, l'effetto Auger è sostanzialmente lo stesso per gli atomi dell'interno del solido e della superficie e può essere utilizzato come metodo assai sensibile per l'analisi chimica delle impurezze che si trovano sulla superficie del campione. Infatti l'energia dell'elettrone emesso dipende unicamente dalla differenza di energia dei livelli coinvolti, caratteristica di ogni specie chimica, e dalla funzione lavoro del solido. Il processo Auger viene indicato specificando: a) lo stato interno da cui viene inizialmente strappato l'elettrone; b) lo stato dell'elettrone più esterno che riempie il ‛buco' formatosi nel processo; c) lo stato dell'elettrone espulso. Così la transizione Auger L23VV è quella in cui un ‛buco' creato nello strato L23 (sovrapposizione degli stati atomici P1/2 e P3/2) è riempito da un elettrone della banda di valenza e l'energia resa disponibile è comunicata a un'altro elettrone della banda di valenza. Nella fig. 16 è riportato lo spettro Auger degli elettroni emessi dal Si in funzione della loro energia; la figura mostra processi del tipo L23VV per energie superiori a 50 eV insieme a processi L1L23V per energie inferiori (L1 corrisponde allo stato S1/2) (v. Chang, 1971).
La fig. 17 mostra invece lo spettro Auger ottenuto da un campione di Si pulito mediante un attacco chimico (etching), in cui, oltre a quello del Si, si vedono chiaramente dei picchi dovuti a impurezze di carbonio e di ossigeno (v. Chang, 1971).
La posizione dei picchi Auger è modificata dalla ridistribuzione di carica in prossimità del sito di adsorbimento dell'impurezza (v. Szalkowski e Somorjai, 1972; v. Somorjai, 1976). Questo chemical shift può essere utilizzato per studiare le proprietà strutturali delle superfici e i meccanismi delle reazioni chimiche superficiali.
Spettroscopia di fotoemissione. - Col termine ‛fotoemissione vengono indicate numerose tecniche basate essenzialmente sulla determinazione dello spettro di energia degli elettroni emessi da un solido per effetto fotoelettrico. Un fascio monocromatico di fotoni del lontano ultravioletto o del campo dei raggi X viene inviato su un solido e vengono misurate le energie cinetiche degli elettroni fotoemessi per mezzo di opportuni analizzatori (simili a quelli usati per le perdite di energia degli elettroni e per la spettroscopia Auger) (v. Feuerbacher e altri, 1978; v. Carlson, 1978; v. Fadley, 1978).
Il processo è schematizzato nella fig. 1 8, nella quale sono indicati alcuni parametri di particolare interesse per la fotoemissione: Evac, EF, Ecin (l'energia cinetica degli elettroni fotoemessi). Nella parte A della figura è riportata, in una direzione dello spazio {k}, l'ipotetica struttura a bande di un solido comprendente uno stato di superficie appena sotto EF. Nella parte B è riportata invece, in ascisse, la densità degli stati elettronici N(E), ossia il numero di stati permessi per intervallo unitario di energia in funzione dell'energia. Ovviamente N(E) può essere calcolato a partire dalla struttura a bande (completa) della fig. 18A. Il fotone di energia ℏω eccita otticamente un elettrone dallo stato pieno A (sotto EF) allo stato vuoto B. La transizione è verticale, nello spazio {k}, essendo il vettore d'onda del fotone (2π/λ) trascurabile rispetto a quello dell'elettrone (dell'ordine di 2π/a). Alcuni elettroni, che non subiscono processi di urto multiplo, arrivano alla superficie con energia superiore a Evac e vengono fotoemessi. Il numero n(E) degli elettroni fotoemessi in funzione della rispettiva energia cinetica può essere misurato dall'analizzatore e dà quella che vien chiamata la curva EDC (Energy Distribution Curve), mostrata nella fig. 18C. La EDC riproduce abbastanza fedelmente la densità degli stati iniziali e può dare informazioni sulla struttura a bande del solido e della sua superficie.
Gli elettroni che non subiscono processi di diffusione multipla provengono da strati appena sotto la superficie (v. fig. 7). Il metodo della fotoemissione è perciò particolarmente sensibile alla struttura elettronica delle superfici.
In generale si usa classificare i processi di fotoemissione a seconda dell'energia dei fotoni utilizzati. Per fotoni di energia compresa fra pochi eV e alcune centinaia di eV si parla di UPS (Ultraviolet Photoemission Spectroscopy). Per fotoni di energia superiore a qualche keV si parla di XPS (X-ray Photoemission Spectroscopy). La XPS è talvolta indicata, in modo alquanto riduttivo, con l'acrostico ESCA (Electron Spectroscopy for Chemical Analysis), per metterne in evidenza le grandi potenzialità per l'analisi chimica.
La UPS è in grado di dare informazioni sulla struttura della banda di valenza dei solidi e sugli stati di superficie associati alla gap principale. La XPS eccita gli elettroni dagli stati più interni degli atomi, le cui energie (poco influenzate dalla superficie) sono rivelate con grande precisione dalla spettroscopia atomica. Essa fornisce pertanto un metodo molto sensibile per analizzare il contenuto di impurezze di una superficie.
I posti vuoti negli stati atomici interni vengono riempiti da elettroni di energia superiore, spesse volte mediante processi Auger. Strutture Auger, anch'esse caratteristiche della specie chimica, accompagnano sempre gli spettri XPS. Un tipico spettro XPS è mostrato nella fig. 19. Le energie degli stati elettronici sono riferite a EF; esse si ottengono sottraendo all'energia ℏω del fotone la somma dell'energia cinetica dell'elettrone fotoemesso e della funzione lavoro.
La fig. 20 mostra invece lo spettro UPS ottenuto nel caso della superficie del Si (111)-2 × 1 (v. Eastman e Grobman, 1972; v. Wagner e Spicer, 1972). Le due curve si riferiscono alla superficie sfaldata e a quella ossidata. La differenza mostra chiaramente uno stato di superficie circa 1 eV sotto il livello di Fermi.
La teoria delle bande permette di calcolare l'energia degli stati elettronici di superficie in funzione del vettore d'onda (di superficie) k. Dato che la componente del vettore d'onda dell'elettrone parallela alla superficie non cambia durante il processo di emissione, è possibile con esperimenti di spettroscopia di fotoemissione a incidenza variabile fare una mappa delle curve di dispersione E(k) di superficie. Infatti, riferendoci per la definizione degli angoli alla fig. 21, possiamo osservare che l'elettrone fotoemesso con energia Ecin ha un vettore d'onda parallelo alla superficie
k∣ ∣ = (2m Ecin)1/2 sen θ/ℏ. (7)
L'energia dello stato da cui l'elettrone è fotoemesso è d'altra parte data (in modulo) da ℏω = Ecin, sicché, variando l'angolo θ, è possibile determinare l'energia di uno stato in funzione di k∣ ∣, ossia la struttura a bande degli stati elettronici di superficie (v. Rowe e altri, 1974). Ciò è mostrato con grande evidenza nella fig. 22, dove è riportata la distribuzione degli stati di superficie del Si (111)-2 × 1, responsabili del picco nella fig. 20, secondo i vari k della zona di Brillouin (v. Himpsel e altri, 1981). Il dispositivo usato per ottenere l'immagine riportata nella figura analizza contemporaneamente tutti i fotoelettroni emessi ai vari angoli e con una data energia, riproducendo la mappa degli stati nella zona di Brillouin superficiale. Si vede che, aumentando l'energia dello stato iniziale, si passa da una distribuzione essenzialmente simmetrica intorno al centro della BZ a due distribuzioni lungo i lati corti della BZ rettangolare.
La flessibilità e la varietà dei metodi della fotoemissione hanno originato numerose altre tecniche, la cui descrizione esula però dagli scopi della presente rassegna.
La spettroscopia di fotoemissione ha ricevuto negli ultimi anni un notevole impulso dalla disponibilità di sorgenti di ‛luce di sincrotrone', che sfruttano la radiazione elettromagnetica emessa dagli elettroni accelerati radialmente sulle orbite degli anelli di accumulazione (originariamente dei sincrotroni). Lo spettro della ‛luce' di sincrotrone si estende in modo continuo dall'infrarosso ai raggi X ed è particolarmente adatto per gli studi di fotoemissione (v. Kunz, 1976; v. Brown, 1974).
c) Diffusione superficiale di molecole e ioni.
Molecole (o atomi) e ioni vengono spesso utilizzati come sonde per studiare la struttura delle superfici. Sono possibili sia processi elastici di diffrazione che processi anelastici, in cui si analizza lo spettro di velocità delle particelle diffuse o l'emissione di raggi X e di elettroni (per effetto Auger) nei processi di neutralizzazione superficiale degli ioni. È possibile inoltre analizzare mediante la spettroscopia di massa gli ioni secondari emessi per urto o desorbiti per effetto termico.
Diffrazione di fasci molecolari. - Gli elettroni utilizzati nella diffrazione LEED, avendo energia di qualche centinaio di eV, interagiscono essenzialmente con i nuclei degli atomi di superficie. Gli elettroni più esterni sono debolmente legati e costituiscono un bersaglio troppo tenue per contribuire apprezzabilmente alla diffusione degli elettroni incidenti. Le misure LEED danno cioè informazioni sulle posizioni dei nuclei atomici.
Un metodo diffrattivo sensibile alla struttura della nube elettronica è invece quello che fa uso della tecnica dei fasci molecolari (v. Engel e Rieder, 1982; v. Boato e Cantini, 1974; v. Celli ed Evans, 1982; v. Cardillo, 1982). Un fascio di molecole viene prodotto mediante un ugello (nozzle) e un diaframma conico (skimmer) e viene inviato sulla superficie da studiare. Se il foro dell'ugello è grande, la distribuzione delle velocità nel fascio è quella delle molecole del gas della sorgente, ossia una distribuzione maxwelliana, che, partendo da zero, presenta un largo massimo dipendente dalla temperatura. Tale distribuzione non è adatta a esperimenti di diffrazione che richiedono fasci pressoché monocromatici. Se però il foro è adeguatamente piccolo, il gas fuoriesce ad alta velocità e inoltre si raffredda espandendosi nel vuoto della camera. Si ottiene così una distribuzione di velocità molto più stretta di quella maxwelliana, con un massimo spostato a velocità più elevate, tanto da rendere il fascio adatto per esperimenti di diffrazione. Vengono usati usualmente fasci di He, Ne, H2, D2, HD, ecc., anche se i risultati più significativi sono stati ottenuti con He. L'energia cinetica delle molecole del fascio può variare da 10 a 200 meV ed è molto più piccola delle energie degli elettroni degli atomi della superficie. Le molecole incidenti interagiscono cioè con un profilo di potenziale come quello riportato schematicamente nella fig. 23, in cui l'ampiezza della corrugazione è molto ridotta rispetto a quella osservata da un elettrone LEFD. Le misure con fasci molecolari sono pertanto più difficili di quelle LEED e spesso sono limitate a superfici con grande ‛rugosità' microscopica, come, ad esempio quelle dei cristalli ionici.
La teoria della diffrazione dei fasci molecolari è simile a quella degli elettroni e fa anch'essa uso dell'eq. (3).
La fig. 24 mostra l'intensità del fascio diffratto secondo i vari angoli nel caso di un fascio di He su una superficie di LiF (100) (v. Boato e Cantini, 1974).
Analisi delle energie delle particelle diffuse. - I fenomeni sono molto diversi a seconda dell'energia delle particelle incidenti. Quando la particella incidente ha energia cinetica molto inferiore all'energia di legame degli atomi di superficie (che è dell'ordine di qualche eV), l'interazione avviene con la superficie come un tutto e lo scambio di energia e di impulso è associato unicamente all'eccitazione delle vibrazioni reticolari di superficie. Il fascio diffuso contiene dei picchi anelastici da cui è possibile risalire alle energie dei fononi superficiali (v. Toennis, 1982). Quando l'energia della particella incidente è invece dell'ordine di qualche centinaio di eV, l'urto avviene in pratica con un solo atomo superficiale che si comporta come una particella libera di massa M. La dinamica è quella dell'urto di due palle da biliardo e la perdita di energia della particella incidente (di massa m) è legata al rapporto μ = M/m e all'angolo di scattering θ (angolo tra le direzioni orientate della particella incidente e di quella diffusa). Con qualche calcolo di meccanica classica si ottiene (v. Suurmeijer e Boers, 1974)
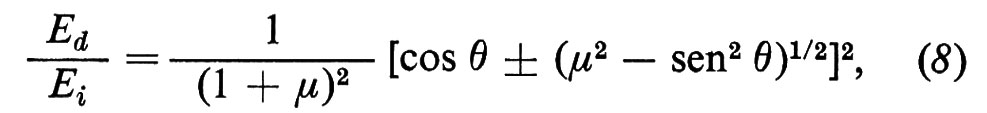
dove Ei ed Ed sono rispettivamente le energie delle particelle incidente e diffusa e il segno + vale per μ > 1. Quando è μ 〈 1, tutti e due i segni sono possibili e l'angolo di scattering non può eccedere il valore arcsenμ. Si vede che una misura di Ei, Ed e θ permette di risalire a μ e cioè alla massa dell'atomo con cui è avvenuta la collisione.
L'esperimento può essere fatto in pratica solo usando, come particelle incidenti, ioni, la cui energia può essere variata (e misurata) mediante opportune differenze di potenziale. La fig. 25 mostra lo spettro delle energie di ioni Ne+ di 300 eV diffusi da una superficie di Ni ricoperta da vari alogeni (v. Brongersma e Mull, 1973). Si vede che il potere risolutivo è tale da rendere possibile una discriminazione tra gli isotopi 35 e 37 del cloro.
Il processo di scattering ora descritto è accompagnato da emissione secondaria di atomi che dà origine a erosione della superficie (sputtering), da fenomeni di incanalamento degli ioni lungo direzioni cristallografiche ben definite (channeling) e da eccitazioni elettroniche con emissione di raggi X e di elettroni (v. White e altri, 1974).
Quando come particella incidente viene usato He+ (o un altro ione di gas nobile) con bassa energia cinetica (~5 eV), lo ione si neutralizza in prossimità della superficie strappando un elettrone da un atomo del solido. Il livello vuoto di He+ è in generale abbastanza profondo, per cui il ‛buco' lasciato nell'atomo di superficie dall'elettrone neutralizzante può essere riempito da un altro elettrone e originare un'emissione di elettroni per effetto Auger. L'analisi dell'energia degli elettroni emessi dà informazioni analoghe a quelle della spettroscopia Auger ed è stata usata per studiare vari processi di chemisorbimento. Questa tecnica viene chiamata spettroscopia da neutralizzazione di ioni (bn Neutralization Spectroscopy, INS) (v. Hagstrum, 1966).
Spettroscopia di massa degli ioni secondari. - La distribuzione delle masse degli ioni secondari emessi rispecchia la composizione chimica della superficie ed è un potente metodo di indagine nella chimica delle superfici. Con gli spettrometri di massa commerciali si ottengono delle sensibilità notevoli (~ 10-6 parti di monostrato) e risoluzione sufficiente a distinguere tutte le specie chimiche. Qualche ambiguità risultante da gruppi atomici con uguale rapporto carica/massa può essere facilmente risolta. Questa tecnica viene usualmente chiamata SIMS (Secondary Ions Mass Spectroscopy) (v. Benninghoven, 1973).
La spettroscopia di massa viene correntemente usata anche per lo studio del desorbimento termico o di quello stimolato dal bombardamento elettronico o dall'assorbimento di radiazione elettromagnetica (v. King, 1975). Questi studi sono particolarmente importanti per la tecnologia degli ultra-alti vuoti.
d) Tecniche di microscopia.
L'osservazione al microscopio (ottico o elettronico) di una superficie permette una valutazione globale atta specialmente a mettere in evidenza difetti su larga scala, contorni di grani, precipitazione di impurezze, ecc. Nel microscopio elettronico a scansione o SEM (Scanning Eleciron Microscope) (v. Johari e Samudra, 1974) la ricostruzione dell'immagine viene fatta elettronicamente, per cui è possibile analizzare il fascetto di elettroni che proviene da uno specifico punto del campione con i metodi della spettroscopia elettronica prima descritti. Se, per esempio, il microscopio è dotato di una sonda Auger, sarà possibile visualizzare la distribuzione di particolari impurezze mettendo in evidenza le regioni dove esse sono state più facilmente chemisorbite o sono precipitate. La risoluzione del SEM è quella degli ordinari microscopi elettronici, ossia dell'ordine di una decina di A.
Dal punto di vista della struttura atomica della superficie sono particolarmente interessanti i microscopi a effetto tunnel e a effetto campo.
Secondo la meccanica quantistica, un elettrone può attraversare barriere di potenziale più alte della sua energia totale. Nella fig. 26A è mostrata una barriera di potenziale triangolare come quella che si ha sulla superficie di un solido cui è applicata una differenza di potenziale. Se F è il campo elettrico uniforme esistente per z > 0, l'energia potenziale dell'elettrone (di carica −q) varia come −qFz. La funzione d'onda di un elettrone di energia E è indicata nella fig. 26B: si vede che essa decade pressoché esponenzialmente tra 0 e d. Si avrà perciò una sensibile probabilità di emissione dell'elettrone per effetto tunnel solo se d è molto piccola, ossia se il campo elettrico applicato è molto grande. La teoria mostra che la probabilità di emissione è proporzionale a (v. Sze, 1973)

dove Δ è l'altezza della barriera di potenziale che l'elettrone deve attraversare, F il campo elettrico applicato e A = 4/3 √-2-m/qℏ. Si vede che la corrente di elettroni emessa per effetto campo cresce molto rapidamente al crescere di F.
Nel microscopio a effetto tunnel, sviluppato recentemente da H. Rohrer e altri (v. Binnig e altri, 1982), una punta metallica molto sottile viene fatta muovere lungo la superficie del campione a piccola distanza dalla stessa. La punta può anche muoversi verticalmente per mantenere costante la distanza punta-campione. Quando un'opportuna ddp è applicata tra campione e punta, si osserva, nel circuito esterno, una corrente dovuta a effetto tunnel. Tale corrente dipende esponenzialmente dal campo elettrico esistente tra la punta e il campione, il quale, a sua volta, è inversamente proporzionale alla distanza della punta. Per tenere costante il valore della corrente mentre la punta si muove lungo la superficie, è necessario spostarla verticalmente in modo da variare la sua distanza dalla superficie stessa. Tali spostamenti, ottenuti in genere mediante dispositivi piezoelettrici, danno una mappa della ‛rugosità' del campione su scala atomica. Il potere risolutivo risulta di circa 5 Å per spostamenti laterali ed è molto migliore per spostamenti verticali, a causa della dipendenza esponenziale espressa dalla (9). Nella fig. 27 è mostrata la mappa di una superficie di Si (111)-7 × 7, ottenuta analizzando tramite calcolatore gli spostamenti verticali della punta (v. Binnig e altri, 1983). Si vedono chiaramente due celle elementari contenenti ciascuna 49 atomi. La direzione [-211] è quella della diagonale lunga di ciascuna cella.
Nel microscopio ionico a effetto campo (Field Ion Microscope, FIM) (v. Müller e Tsong, 1969) è il campione da studiare a essere sagomato a forma di punta. Ad esso viene applicata una forte ddp, con la punta positiva rispetto a uno schermo fluorescente posto ad alcuni centimetri di distanza. In queste condizioni non si ha emissione di elettroni, ma il campo in prossimità della punta (5 ÷ 10 Å) è così intenso da ionizzare degli atomi, in generale di He, introdotti appositamente nella camera. Gli ioni positivi così formati vengono respinti dalla punta e pervengono sullo schermo fluorescente, dove si forma una specie di immagine che riproduce le variazioni laterali del campo elettrico in prossimità della superficie. La risoluzione è di 2 ÷ 4 Å e il metodo è in grado di dare un'immagine dei singoli atomi.
5. Modelli di superfici.
La molteplicità e la versatilità delle tecniche sperimentali descritte nel capitolo precedente hanno permesso negli anni settanta e ottanta uno sviluppo rigoglioso dell'attività di ricerca nel campo della fisica delle superfici. Lo scopo principale di questi studi è quello di ricavare dei modelli per la struttura atomica, elettronica e vibrazionale di una data superficie e di utilizzarli per la risoluzione dei molteplici problemi, anche tecnologici, delineati nel cap. 3. Le tecniche descritte forniscono però il più delle volte risposte parziali ai vari problemi ed è necessario un confronto dettagliato con la teoria se si vogliono ricavare modelli realistici delle superfici.
Nel caso dei semiconduttori, la struttura elettronica delle superfici può essere calcolata nello schema della teoria a una particella di Hartree-Fock, più nota come ‛teoria delle bande'. I risultati vengono spesso presentati tramite la densità locale degli stati (Local Density of States, LDS), che è particolarmente adatta a mettere in evidenza la distribuzione elettronica superficiale.
La densità totale degli stati ρtot(E), ossia il numero di stati presenti in un intervallo unitario di energia intorno all'energia E, può essere scritta come (v. Forstmann, 1978)
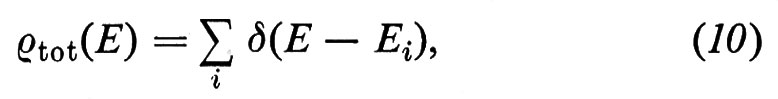
dove δ è la funzione di Dirac e la somma è fatta su tutti gli stati di energia Ei. Sfruttando la proprietà della δ di Dirac, ∫ δ(x) dx = 1, si vede immediatamente che
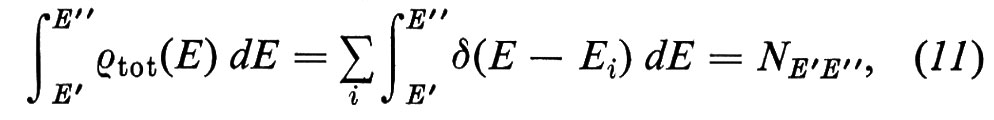
dove NE′E′′ è il numero di stati compresi tra le energie E′ ed E′′. La ρtot(E) soddisfa perciò alla definizione di densità degli stati.
La funzione
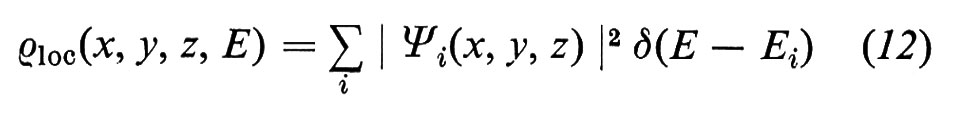
pesa la densità degli stati con la probabilità ∣ Ψi(x, y, z) ∣2 di trovare un elettrone nella posizione x, y, z ed è la LDS cercata. La sua utilità nello studio delle superfici risulta evidente dall'esempio seguente.
Nella fig. 28 è mostrata la disposizione degli atomi nel piano (111) della superficie del Si non ricostruita. I legami sono indicati con dei bastoncini. Nella fig. 29 è riportata la distribuzione di carica nel piano (-110) normale alla superficie (111), per due stati a energie +0,5 e −8,5 eV rispetto alla cima della banda di valenza, calcolata da Marvin L. Cohen e collaboratori con il metodo dello pseudo-potenziale (v. Schlüter e altri, 1975). Si vede chiaramente che nello stato profondo l'elettrone è distribuito pressoché sfericamente intorno all'atomo di superficie (stato s), mentre per lo stato nella gap si ha una distribuzione di carica che protrude dalla superficie nei dangling bonds (stato pz).
I calcoli, non necessariamente limitati alla tecnica dello pseudopotenziale, vengono in generale effettuati su complessi (slabs) di piani atomici infiniti (da 12 a 30), separati da spazi vuoti sufficientemente ampi da evitare l'interazione tra le superfici affacciate e disposti periodicamente. La periodicità imposta agli slabs permette una convergenza più rapida delle soluzioni senza alterare la sostanza del problema (v. Alldredge e Kleinman, 1972). Alcuni parametri vengono determinati in modo da riprodurre le proprietà del bulk (per es. la LDS) nei piani più interni di ciascuno slab.
In linea di principio calcoli come quelli illustrati nella fig. 29, ripetuti per tutte le possibili geometrie di superficie, permetterebbero di prevedere, per mezzo dei teoremi variazionali della meccanica quantistica, la ricostruzione superficiale. Metodi variazionali sono stati tentati nell'ambito di alcuni modelli, ma non hanno dato risultati conclusivi (v. Chadi, 1978). Può essere perciò interessante analizzare come dall'interconnessione tra i vari risultati sperimentali e il calcolo teorico sia possibile giungere in qualche caso a un modello affidabile di ricostruzione.
Ci riferiremo, a titolo di esempio, alla superficie di sfaldatura del silicio Si (111)-2 × 1 (e a quella del germanio, strettamente affine), per la quale, nel cap. 4, sono stati riportati alcuni importanti risultati sperimentali. Si vede dalla fig. 28 che nella superficie ideale è presente un dangling bond per atomo di superficie e che pertanto, essendoci un solo elettrone per legame (l'altro è rimasto associato nella sfaldatura all'altra metà del solido), la superficie dovrebbe avere carattere metallico. In queste condizioni il sistema è instabile, specie se, come in superficie, gli atomi hanno una certa libertà di spostamento. Infatti la trasformazione metallo-semiconduttore, con l'apertura di una gap, riduce in genere l'energia elettronica totale del sistema abbassando l'energia dei livelli pieni e aumentando l'energia dei livelli vuoti (v. Tosatti, 1975). Questa transizione è evidenziata dalla struttura 2 × 1 (LEED: v. fig. 9), nella quale la cella elementare contiene due atomi e perciò un numero pari di elettroni nei dangling bonds, come richiesto per un isolante o per un semiconduttore.
L'esistenza di due stati (bande) elettronici superficiali, uno pieno e l'altro vuoto, è messa in evidenza dallo spettro di riflettività differenziale della fig. 12 e di EELS della fig. 15, che mostrano appunto nel picco a 0,45 eV (e a 0,52 eV per EELS) l'esistenza di una gap. Le misure ottiche mettono anche in evidenza una grande anisotropia della superficie, mostrando che solo luce polarizzata con il vettore elettrico nella direzione [1-10] della superficie ricostruita può essere assorbita nella transizione associata ai dangling bonds.
Poiché lo stato di superficie osservato in fotoemissione è situato circa 1 eV sotto la cima della banda di valenza (v. fig. 20), sembrava improbabile che esso fosse lo stato iniziale della transizione ottica a 0,45 eV. Le misure di fotoemissione risolta angolarmente (v. fig. 22) mostrano però che la struttura del picco della fig. 20 è complessa e che esiste, a energie corrispondenti alla cima della banda di valenza, una distribuzione di stati (attorno al lato corto della BZ rettangolare della superficie 2 × 1) che costituiscono gli stati di partenza della transizione ottica osservata.
In conclusione, gli stati di superficie del Si (111)-2 × 1 hanno una distribuzione in energia come quella indicata schematicamente nella fig. 30. L'incurvamento delle bande del bulk, riportato nella parte sinistra della figura, è dovuto al potenziale macroscopico causato dall'accumulo di carica negativa negli stati di superficie.
Dal punto di vista della ricostruzione si può osservare che la grande anisotropia della superficie, messa in evidenza dalle misure ottiche, suggerisce un modello anisotropo con strutture di atomi allineati in qualche modo nelle direzioni [110]. Tale modello è stato proposto da K. C. Pandey (v., 1981) nel 1981 ed è quello ormai generalmente accettato.
Esso è mostrato schematicamente per il caso del Ge (111)-2 × 1 (v. Northrup e Cohen, 1983), strettamente analogo a quello del Si, nella fig. 31. Si vede chiaramente che gli atomi della superficie (111) si dispongono in catene lungo le direzioni [1-10] separate da avvallamenti.
Gli atomi del germanio e del silicio tetravalenti sono caratterizzati da quattro legami tetraedrici di tipo sp3. La figura mostra che gli atomi delle catene hanno solo tre legami ciascuno. Il legame mancante (dangling bond) è utilizzato per un ulteriore legame (di tipo π, simile a quello che si riscontra nella molecola del benzene) tra gli atomi della catena stessa. In questo modello la transizione ottica corrisponderebbe alla rottura del legame π o a una transizione tra gli stati di bonding e di antibonding della catena.
Il modello di Pandey rappresenta una ricostruzione del tipo 2 × 1, in quanto la cella elementare è raddoppiata nella direzione perpendicolare alle catene. Esso spiega anche la grande dispersione delle energie degli stati di superficie, osservata nella fotoemissione risolta angolarmente ed evidente nella fig. 30, a causa della grande interazione tra gli atomi delle catene assai prossimi tra loro.
Dalla fig. 31 risulta che la ricostruzione 2 × 1 nel modello a catene richiede la rottura di alcuni legami, un processo apparentemente molto improbabile, dato che l'energia di legame è dell'ordine di qualche eV. Calcoli autoconsistenti nello schema della LDS hanno mostrato però che la nube elettronica si deforma con continuità passando dalla superficie ideale a quella ricostruita e che l'energia di attivazione è inferiore a 0,1 eV/atomo (v. Northrup e Cohen, 1982).
Nei metalli l'approssimazione a un elettrone è in generale inadeguata e i calcoli sono più complessi. Un modello che ha dato buoni risultati per il calcolo della funzione lavoro è il cosiddetto jellium. In tale modello si assume che gli elettroni costituiscano un gas di particelle interagenti (gas di Fermi), che si muovono in un mezzo neutralizzante di cariche positive distribuite uniformemente. Il sistema è caratterizzato da un unico parametro: la densità degli elettroni. La superficie é simulata assumendo che la distribuzione di cariche positive si interrompa bruscamente e calcolando la risposta del gas di elettroni a tale interruzione. Si trovano oscillazioni quantistiche (oscillazioni di Friedel) nella densità degli elettroni in prossimità della superficie (v. Appelbaum e Haman, 1976; v. Garcia Moliner e Flores, 1979). Modelli più accurati, che tengono conto della struttura discontinua del solido e della superficie, sono stati sviluppati specie in connessione con l'emissione fotoelettrica (v. Willis e Feuerbacher, 1978).
Si vede da questi esempi che la formulazione di un modello teorico per una superficie ricostruita non è un procedimento semplice e richiede l'uso e il confronto di molteplici tecniche e di raffinati metodi teorici. In casi fortunati il LEED dinamico ha dato dei risultati affidabili, che hanno permesso di ricavare modelli non controversi di ricostruzione. Alcune superfici, tra cui la 7 × 7 del silicio, hanno però sfidato finora gli sforzi e la fantasia dei ricercatori. Il loro studio ha in ogni caso stimolato una grande attività di ricerca e un avanzamento generale delle nostre conoscenze di fisica delle superfici.
Bibliografia.
Alldredge, G., Kleinman, L., New method of calculating bulk and surface states in thin films, in ‟Physical review letters", 1972, XXVIII, pp. 1264-1268.
Appelbaum, J.A., Haman, D.R., The electronic structure of solid surfaces, in ‟Review of modern physics", 1976, XLVIII, pp. 479-496.
Aspnes, D.E., Spectroscopy ellipsometry of solids, in Optical properties of solids. New developments (a cura di B.O. Seraphin), Amsterdam-New York 1976, pp. 799 ss.
Benninghoven, A., Surface investigation of solids by the statistical method of secondary ion mass spectroscopy (SIMS), in ‟Surface science", 1973, XXXV, pp. 427-457.
Binnig, G., Rohrer, H., Gerber, C., Weibel, E., Surface studies by scanning tunneling microscopy, in ‟Physical review letters", 1982, IL, pp. 57-61.
Binnig, G., Rohrer, H., Gerber, C., Weibel, E., 7 × 7 reconstruction on Si (111) resolved in real space, in ‟Physical review letters", 1983, L, pp. 120-123.
Boato, G., Cantini, P., Experiments on scattering of light atomic beams from crystal surfaces, in Aspetti dinamici della fisica delle superfici. Rendiconti della Scuola Internazionale di Fisica ‛E. Fermi', LVIII corso, Bologna 1974, pp. 707-735.
Brongserma, H.H., Mull, P.M., Analysis of the outermost atomic layer of a surface by low-energy ion scattering, in ‟Surface science", 1973, XXXV, pp. 393-412.
Brown, F.C., Ultraviolet spectroscopy of solids with the use of synchrotron radiation, in Solid state physics (a cura di H. Ehrenreich, F. Seitz e D. Turnbull), vol. XXIX, New York-London 1974, pp. 1-73.
Cardillo, M. J., He diffraction from semiconductor surfaces, in Dynamics of gas-surface interaction (a cura di G. Benedek e U. Valbusa), Berlin-New York 1982, pp. 40 ss.
Carlson, T.A., Photoelectron and Auger spectroscopy, New York-London 1978.
Carnevali, P., Selloni, A., Microscopia a effetto tunnel, in ‟Note di informatica, IBM", 1984, VII, pp. 3-20.
Celli, V., Evans, D., Theory of atom-surface scattering, in Dynamics of gas-surface interaction (a cura di G. Benedek e U. Valbus), Berlin-New York 1982, pp. 2 ss.
Chabal, Y.J., Raghavachari, K., Surface infrared study of Si (100) - (2 × 1) H, in ‟Physical review letters", 1984, LIII, pp. 282-285.
Chadi, D.J., Energy-minimization approach to the atomic geometry of semiconductor surface, in ‟Physical review letteres", 1978, XLI, pp. 1062-1065.
Chang, C.C., Auger electron spectroscopy, in ‟Surface science", 1971, XXV, pp. 53-79.
Chiaradia, P., Chiarotti, G., Nannarone, S., Sassaroli, P., Surface states on Si (111) 2 × 1 detected by external reflectivity, in ‟Solid state communications", 1978, XXVI, pp. 813-815.
Chiaradia, P., Cricenti, A., Selci, S., Chiarotti, G., Differential reflectivity of Si (111) 2 × 1 surface with polarized light. A test for surface structure, in ‟Physical review letters", 1984, LII, pp. 1145-1147.
Chiarotti, G., Del Signore, G., Nannarone, S., Optical detection of surface states on cleaved (111) surfaces of Ge, in ‟Physical review letters", 1968, XXI, pp. 1170-1172.
Chiarotti, G., Nannarone, S., Pastore, R., Chiaradia, P., Optical absorption of surface states in ultrahigh vacuum cleaved (111) surfaces of Ge and Si, in ‟Physical review, B", 1971, IV, pp. 3398-3402.
Citrin, P.H., Eisenberger, P., Hewitt, B.M., Extended X-ray-absorption fine structure of surface atoms on single-crystal substrates: iodine adsorbed on Ag (111), in ‟Physical review letters", 1978, XLI, pp. 309-312.
Comin, F., Rowe, J.E., Citrin, P.H., Structure and nucleation mechanism of nickel silicide on Si (111) derived from surface extended X-ray-absorption fine structure, in ‟Physics review letters", 1983, LI, pp. 2402-2405.
Eastman, D.E., Grobman, W.D., Photoemission densities of intrinsic surface states for Si, Ge and GaAs, in ‟Physical review letters", 1972, XXVIII, pp. 1378-1381.
Engel, T., Rieder, K.H., Structural studies of surface with atomic and molecular beam diffraction, in Structural studies of surfaces. Springer tracts in modern physics, Berlin-New York 1982, vol. XCI, pp. 5 ss.
Fadley, C.S., Basic concepts of X-ray photoelectron spectroscopy, in Electron spectroscopy (a cura di C.R. Brundle e A.D. Baker), London-New York 1978, vol. II, pp. 2 ss.
Feuerbacher, B., Fitton, B., Willis, R.F. (a cura di), Photoemission and the electronic properties of surfaces, New York 1978.
Forstmann, F., Quantum theory of surfaces and interfaces, in Theory of imperfect crystalline solids, International Atomic Energy Agency, Wien 1981, pp. 511 ss.
Forstmann, F., Electron states at clean surfaces, in Photoemission and the electronic properties of surfaces (a cura di B. Feuerbacher, B. Fitton e R.F. Willis), New York 1978, pp. 193 ss.
Froitzheim, H., Electron loss spectroscopy, in Electron spectroscopy for surface analysis (a cur di H. Ibach), Berlin-New York 1977, pp. 205 ss.
Froitzheim, H., Ibach, H., Mills, D.L., Surface optical constants of silicon and germanium derived from electron-energy-loss spectroscopy, in ‟Physical review, B", 1975, XI, pp. 4980-4988.
García Moliner, F., Flores, F., Introduction to the theory of solid surfaces, London-New York 1979, pp. 146 ss.
Hagstrum, H.D., Ion-neutralization spectroscopy of solids and solid surfaces, in ‟Physical review", 1966, CL, pp. 495-515.
Harrick, N. J., Beckmann, K.H., Internal reflection spectroscopy, in Characterization of solid surfaces (a cura di P. F. Kane e G.B. Larrabee), New York 1974, pp. 215 ss.
Heinz, K., Muller, K., LEED intensities experimental progress and new possibilities of surface structure determination, in Structural studies of surface, Berlin-New York 1982, vol. XCI, pp. 1 ss.
Henzler, H., LEED-investigation of step arrays on cleaved germanium (111) surfaces, in ‟Surface science", 1970, XIX, pp. 159-171.
Himpsel, F.J., Heimann, P., Eastman, D.E., Surface states on Si (111) - (2 × 1), in ‟Physical review, B", 1981, XXIV, pp. 2003-2008.
Ibach, H. (a cura di), Electron spectroscopy for surface analysis, Berlin-New York 1977.
Johari, O., Samudra, A.V., Scanning electron microscope, in Characterization of solid surfaces (a cura di P.F. Kane e G.B. Larrabee), New York, 1974, pp. 157 ss.
Jona, F., Low energy electron diffraction (LEED) spectra: Aluminum, in ‟IBM journal of research and development", 1970, XIV, pp. 444-452.
King, D.A., Thermal desorption from metal surfaces: a review, in ‟Surface science", 1975, XLVII, pp. 384-402.
Kunz, C., Synchrotron radiation as a source for photoemission, in Photoemission and the electronic properties of surfaces (a cura di B. Feuerbacher, B. Fitton e R.F. Willis), New York 1976, pp. 501 ss.
McIntyre, J.D.E., Aspnes, D.E., Differential reflection spectroscopy of very thin surface films, in ‟Surface science", 1971, XXIV, pp. 417-434.
Many, A., Electrical transport in the space charge region, in Surface science, International Atomic Energy Agency, Wien 1975, vol. I, pp. 4787 ss.
Margaritondo, G., Microscopic investigations of semiconductor surfaces, in ‟Solid state electronics", 1983, XXIV, pp. 499-513.
Müller, E.W., Tsong, T.T., Field ion microscopy. Principles and applications, Amsterdam 1969.
Northrup, J.E., Cohen, M.L., Reconstruction mechanism and surface-state dispersion for Si (111) - (2 × 1), in ‟Physical review letters", 1982, IL, pp. 1349-1352.
Northrup, J.E., Cohen, M. L., Atomic geometry and surface-state spectrum for Ge (111) - (2 × 1), in ‟Physical review, B", 1983, XXVII, pp. 6553-6556.
Pandey, K.C., New π-bonded chain model for Si (111) - (2 × 1), surface, in ‟Physical review letters", 1981, XLVII, pp. 1913-1917.
Pendry, J.B., Low energy electron diffraction, London-New York 1974, pp. 220 ss.
Raether, H., Solid state excitations by electrons, in Springer tracts in modern physics, Berlin-New York 1965, vol. XXVIII, pp. 84 ss.
Rivière, J. C., Auger electron spectroscopy, in ‟Contemporary physics", 1973, XIV, pp. 513-539.
Rowe, J. E., Ibach, H., Froitzheim, H., Photoemission and energy loss spectroscopy on semiconductor surfaces, in ‟Surface science", 1975, XLVIII, pp. 44-58.
Rowe, J. E., Traum, M. M., Smith, N. V., Measurement of the angle of dangling-bond photoemission from cleaved silicon, in ‟Physical review letters", 1974, XXXIII, pp. 1333-1335.
Schlüter, M., Chelikowsky, J. R., Louie, S. G., Cohen, M. L., Self-consistent pseudopotential calculations for Si (111) surfaces: unreconstructed (1×1) and reconstructed (2×1) model structures, in ‟Physical review, B", 1975, XII, pp. 4200-4210.
Selci, S., Chiaradia, P., Ciccacci, F., Cricenti, A., Chiarotti, G., Polarization-dependent reflectivity of Si (111)-(2×1) surface above the gap, in ‟Physical review, B", 1985, XXXI, pp. 4096-4098.
Somorjai, G. A., The structure and thermodynamics of clean surfaces, in Solid state chemistry (a cura di N. B. Hannay), New York-London 1976, vol. VI A, pp. 1 ss.
Suurmeijer, E. P. Th. M., Boers, A. L., Low-energy ion reflection from metal surfaces, in ‟Surface science", 1974, XLIII, pp. 309-352.
Szalkowsky, F. J., Somorjai, G. A., Auger electron spectroscopy investigations of the surface chemical composition of vanadium, the vanadium oxides, and oxidized vanadium: chemical shift and peak intensity analysis, in ‟Journal of chemical physics", 1972, LVI, pp. 6097-6103.
Sze, S. M., Fisica dei dispositivi a semiconduttore, Milano 1973.
Toennis, J. P., Phonon interactions in atom scattering from surfaces, in Dynamics of gas-surface interactions (a cura di G. Benedek e U. Valbusa), Berlin-New York 1982, pp. 208 ss.
Tosatti, E., Electron superstructures of semiconductor surfaces and layered transition metal compounds, in Festkörperprobleme (a cura di H. J. Queisser), Braunschweig 1975, pp. 113 ss.
Tosatti, E., Semiconductor. Surface rec0nstruction, in Proceedings of the 13th international conference on the physics of semiconductors, Roma 1976, pp. 21 ss.
Voorhoeve, R. J. H., Molecular beam deposition of solid surfaces, in Solid state chemistry (a cura di N. B. Hannay), New York-London 1976, vol. VI A, pp. 241 ss.
Wagner, L. F., Spicer, W. E., Observation of a band of silicon surface states containing one electron per surface atom, in ‟Physical review letters", 1972, XXVIII, pp. 1381-1384.
White. C. W., Simms, D. L., Talk, N. H., Surface composition by analysis at neutral and ion impact radiation, in Characterization of solid surfaces (a cura di P. F. Kane e G. B. Larrabee), New York 1974, pp. 215 ss.
Willis, R. F., Feuerbacher, B., Photoemission from metals, in Photoemission and the electronic properties of surfaces (a cura di B. Feuerbacher, B. Fitton e R. F. Willis), New York 1978, pp. 281 ss.
Chimica delle superfici di Alessandro Cimino
SOMMARIO: 1. Introduzione. □ 2. Reattività della superficie: a) natura del legame superficie-molecola; b) il ‛sito' superficiale. □ 3. La superficie come sede di siti a più funzioni: a) stati ionici superficiali; b) struttura e proprietà di siti in solidi reali; c) proprietà acide e basiche; d) allumina y e zeoliti: due casi tipici nella chimica delle superfici; e) chimica dei clusters e chimica delle superfici. □ 4. Termodinamica delle superfici e composizione superficiale. □ 5. Esempi di applicazioni di tecniche spettroscopiche a problemi di superficie: a) spettroscopia infrarossa; b) spettroscopia fotoelettronica; c) risonanza di spin elettronico (ESR). □ 6. Nota conclusiva. □ Bibliografia.
1. Introduzione.
La chimica delle superfici ha per oggetto di studio l'insieme dei fenomeni chimici che hanno luogo sulla ‛superficie' di uno stato condensato, in particolare di un solido. Gli atomi (o ioni) presenti su una superficie sono intrinsecamente in uno stato diverso da quelli che si trovano all'interno della fase condensata, dato che solo una parte delle loro valenze è saturata da atomi vicini. Questo stato genera negli atomi superficiali una reattività e una capacità di comportamento chimico particolari, che giustificano l'esistenza di una chimica delle superfici.
Tra le superfici, quelle che delimitano i solidi sono di gran lunga più importanti sia per gli aspetti teorici, date le differenti configurazioni che possono realizzarsi, sia per gli aspetti applicativi, data la varietà di materiali e di fenomeni che coinvolgono la superficie di un solido. Basterà citare tra questi ultimi la catalisi eterogenea, la corrosione, l'elettrochimica degli elettrodi, l'adesione e la lubrificazione. Importanti settori applicativi, oltre a quelli citati, coinvolgono fenomeni di superficie, per esempio l'utilizzazione dell'energia solare, la fisica dei semiconduttori, ecc.
È opportuno chiarire che cosa si debba intendere per superficie. Da quanto premesso si potrebbe supporre che una chimica delle superfici distinguibile da quella che si occupa dell'interno della massa dei materiali o delle soluzioni possa riguardare solo gli atomi esterni, dato il rapido smorzamento delle forze attrattive e repulsive. Devono peraltro farsi le seguenti considerazioni: a) la geometria della disposizione degli atomi su una superficie non è sempre tale da ‛nascondere' gli atomi sottostanti; vi sono facce cristalline (v. fig. 1) nelle quali gli atomi sottostanti sono più o meno ‛scoperti', e anche questi ultimi devono quindi essere considerati potenzialmente partecipi e facenti parte della ‛superficie'; b) la partecipazione di atomi sottostanti è accentuata da possibili ‛ricostruzioni'; c) in molti fenomeni si deve tenere conto dei fenomeni elettronici che modificano la struttura elettronica alla superficie, e quindi la reattività degli atomi esterni. Le modifiche della struttura elettronica possono risentirsi a distanza e dipendere quindi da strati sottostanti, la cui natura e composizione hanno pertanto influenza sulla chimica di superficie.
Si può quindi comprendere che il campo di studio della chimica delle superfici, a volte chiamata in modo suggestivo ‛chimica a due dimensioni', non ha un confine geometrico netto. L'esigenza di includere strati più o meno profondi dipende in realtà dal fenomeno studiato; ciò si riflette nella diversità e nell'ampiezza degli argomenti trattati, e nelle metodologie sperimentali. In genere si tende oggi a includere nella chimica delle superfici i processi che coinvolgono gli ultimi strati di un cristallo. È anche da notare che non si ha un passaggio netto e discontinuo tra fenomeni che interessano solo gli atomi della superficie (per es. nel vuoto) e quelli che avvengono in presenza di molecole esterne (per es. nell'adsorbimento). Pertanto la chimica delle superfici studia i processi di adsorbimento, indicativi della reattività superficiale e che costituiscono gli stati iniziali dei processi di corrosione e della catalisi eterogenea, la natura delle specie presenti sulle superfici e le loro reattività. La chimica delle superfici si occupa, invece, marginalmente dei fenomeni fisici di condensazione, trattati in genere dalla termodinamica delle interfasi, anche se questi fenomeni sono di notevole importanza, dal punto di vista tecnico, nella caratterizzazione della ‛tessitura' dei solidi (estensione della superficie, porosità, ecc.). In anni recenti l'attenzione dei ricercatori si è andata sempre più concentrando sul fenomeno dell'arricchimento superficiale in solidi, per le importanti conseguenze che esso ha sulla reattività della superficie.
È da tener presente che vanno prese in considerazione anche le superfici interne, quali quelle esistenti dentro pori accessibili, e che l'accessibilità dipende dalle specie reagenti. Pori molto piccoli escludono molecole ingombranti, per cui ciò che è superficie per una reazione può non esserlo per altre. Un caso estremo è offerto dalle zeoliti, una classe di solidi cui appartengono composti naturali e artificiali, di particolare interesse applicativo nella catalisi. Nelle zeoliti non si hanno ‛pori' nel senso comune, ma canali insiti nella struttura, accessibili a molecole piccole. La chimica delle superfici prende in tal caso in esame anche la struttura e la reattività delle superfici interne della cavità, senza confini netti tra chimica delle superfici e chimica dello stato solido.
Il presente articolo dà un panorama necessariamente sommario dei problemi che rientrano nel settore della chimica delle superfici.
2. Reattività della superficie.
a) Natura del legame superficie-molecola.
L'esistenza di atomi o ioni non saturi coordinativamente consente la formazione di legami chimici tra la superficie e una specie appartenente a una fase esterna. Per brevità indicheremo con il nome generico di ‛molecola' l'unità (molecola, atomo o ione) costitutiva della fase esterna. Le due entità che si legano sono quindi la molecola A e la superficie S. Quest'ultima viene a svolgere il ruolo di un secondo atomo B nella formazione di una molecola AB. Non è necessario supporre che sia un singolo atomo appartenente alla superficie a legare la molecola adsorbita: si può infatti pensare che si tratti di un particolare gruppo di atomi Bn della superficie stessa (v. È b). A prescindere dal modello, il legame che si forma è un vero legame chimico, come risulta per esempio dalle energie in gioco nella formazione del legame (v. catalisi eterogenea, cap. 2 e fig. 1) e dalle indicazioni spettroscopiche (v. cap. 5, È a). Si comprende quindi che il legame A−S potrà essere trattato in una varietà di modi in dipendenza dal sistema e dagli obiettivi proposti. Le diverse trattazioni riflettono da un lato l'esigenza di adottare un modello sufficientemente realistico, e dall'altro l'impossibilità di trattare in maniera rigorosa modelli troppo complessi.
Un problema di base è quello della descrizione teorica del legame stesso, in funzione sia della struttura elettronica della molecola, sia di quella della superficie del solido. Riguardo a quest'ultima, l'articolo superfici: Fisica delle, suppl., presenta una trattazione da cui emerge la complessità della descrizione di una superficie reale: pertanto, quando si tenta di applicare i principi impiegati nella teoria del legame chimico al caso molecola-superficie si incontrano difficoltà non sormontabili se non tramite semplificazioni, che rischiano però di non tener conto di importanti caratteristiche del legame; è quindi necessario unire l'intuizione chimica ai criteri di semplificazione.
Si può tentare di descrivere il legame considerando due casi limite: a) legame ionico con trasferimento elettronico; b) legame covalente. Il caso a) coinvolgerà parametri relativamente semplici del solido e della molecola: energia di estrazione degli elettroni e livello di Fermi per il solido, elettroaffinità ed energia di ionizzazione per la molecola. Si può quindi descrivere il legame come un trasferimento elettronico alla molecola, o dalla molecola. Questo processo conduce alla formazione di un doppio strato elettrico che blocca l'ulteriore passaggio di carica. Siffatta teoria, sviluppata intorno al 1950 e detta teoria dello ‛strato esterno' (boundary layer in inglese e Randschicht in tedesco), dava risultati qualitativamente corretti per alcuni sistemi: per esempio rendeva conto della tendenza di ZnO ad adsorbire ossigeno e prevedeva correttamente limiti di copertura molto bassi. Un breve esame della teoria dello strato esterno è svolto nell'articolo catalisi eterogenea, cap. 2; per trattazioni più ampie v. García-Moliner, 1968; v. Morrison, 1977; per un riepilogo sommario di questa e di altre trattazioni v. anche Cimino e Carrà, 1980.
Per quanto riguarda il caso b) si ricorda che quasi contemporaneamente alle trattazioni sopra citate venivano svolti tentativi, di tipo semiempirico, di descrizione del legame, che tenevano conto dei risultati della meccanica quantistica per legami chimici semplici e delle proprietà collettive dei solidi. Tra questi si ricorda la cosiddetta ‛teoria elettronica' della catalisi e dell'adsorbimento, sviluppata principalmente da Wolkenstein (v., 1963) in Unione Sovietica (v. catalisi eterogenea, cap. 2), che si applicava in particolare al comportamento dei semiconduttori. Questa e altre trattazioni, pure basate su proprietà collettive, hanno avuto l'indubbio merito di aver posto l'attenzione sugli aspetti elettronici dell'interazione e sul ruolo del solido, in particolare sulla sua struttura elettronica di superficie. Esse hanno fornito però più un quadro per la razionalizzazione a posteriori che non una vera teoria di base.
La difficoltà di applicazione di queste teorie a casi reali e il progresso registrato nella descrizione del legame chimico nei composti di coordinazione in chimica inorganica hanno indotto alcuni studiosi a sviluppare una trattazione di tipo diverso, basata non sulle proprietà collettive del solido, ma proprio sulle teorie dei composti di coordinazione. Il legame molecola-superficie veniva cioè trattato come parte di un ‛composto' o ‛complesso' superficiale, e si è pertanto designata questa trattazione come ‛localizzata'. E da citare in proposito l'opera di Dowden (v., 1971), che metteva in luce la possibilità di stabilizzazione di particolari simmetrie sulla superficie, in accordo con la teoria del campo dei leganti. Veniva così correlata la stabilità relativa dei complessi superficiali con la loro reattività.
La validità dei modelli basati su proprietà collettive dipende peraltro dall'intensità dell'interazione molecola-superficie e quindi non può essere invocata a priori la superiorità di un metodo sull'altro: si tratta di approssimazioni da valutare caso per caso. Si può riassumere e illustrare in modo schematico il significato delle diverse approssimazioni con l'aiuto della fig. 2. Si abbia nella molecola o atomo A un livello come in (a) e nel solido una struttura a bande come in (e), che rappresenta la densità degli stati in superficie (D. S. Sup.) in funzione dell'energia E. Per interazione tra queste configurazioni, in accordo con la teoria degli orbitali molecolari, si avrà una perturbazione di entità diversa da caso a caso. In (b) si ha debole interazione del livello di A e la densità degli stati (D. S.) è praticamente ancora data da un livello. In (d) si ha una forte interazione, in (c) una situazione intermedia. Nel caso (b) l'interazione molecola-superficie può essere ancora descritta sulla base delle proprietà di partenza di A, da un lato, e sulla base della struttura collettiva del solido, dall'altro. Nel caso (d) è chiaro che la forte perturbazione non permette una descrizione semplificata.
Un breve cenno infine alle trattazioni del problema del legame con una superficie, sulla base di considerazioni teoriche più rigorose, sviluppate soprattutto in anni recenti con un'impostazione che rientra nella chimica teorica e nella fisica dei solidi. Si è cercato, per esempio, di definire il ruolo degli ‛stati superficiali', dei quali un caso (dangling bonds) è stato esposto nell'articolo superfici: Fisica delle, suppl. Questi stati possono essere importanti in alcuni fenomeni - per esempio possono partecipare all'adsorbimento - ma non necessariamente in altri, quali la catalisi, proprio perché facilmente e stabilmente saturabili. D'altra parte non è essenziale che gli stati superficiali localizzati preesistano: essi possono formarsi proprio a seguito dell'interazione molecola-superficie, in modo analogo agli orbitali molecolari leganti, creati appunto nella formazione del legame chimico. Per un'esposizione delle trattazioni teoriche si veda, ad esempio, il libro curato da Rhodin ed Ertl (v., 1979) e i riferimenti ivi contenuti.
È istruttivo come commento finale citare alcune parole della prefazione di Rhodin ed Ertl: ‟Un testo che tratti il campo del legame chimico di superficie in questo tempo è in un certo senso più un rendiconto di successi potenziali che non una documentazione di risultati raggiunti". Una dichiarazione tuttora valida, che indica un futuro campo di sviluppo.
b) Il ‛sito' superficiale.
Dal punto di vista chimico è di particolare interesse chiarire il concetto di ‛sito', che nel caso dell'adsorbimento svolge la funzione di punto di ancoraggio della molecola alla superficie. Sono state richiamate nel È a diverse trattazioni, localizzate e non, a seconda del tipo di approssimazione seguita nel descrivere la struttura elettronica del solido. In entrambi i tipi di modelli, una volta formato, il legame coinvolge direttamente un numero limitato di atomi superficiali, nel caso più semplice uno solo. Con le nuove tecniche sperimentali, in particolare quelle spettroscopiche, si è potuto mettere in evidenza che per una stessa molecola il legame può essere stabilito con uno, o due, o più atomi della superficie a seconda della natura della superficie, della copertura e della temperatura. Il concetto di ‛sito' non è quindi necessariamente coincidente con quello di ‛atomo superficiale'. A volte si intende per sito l'insieme degli atomi superficiali interessati al legame. Alcuni autori indicano con ‛sito' ogni singolo atomo partecipante al legame, e nel caso sopra citato si dirà che l'adsorbimento richiede uno, due o più ‛siti'. In catalisi si parla di ‛centro attivo' per definire il sito o l'insieme dei siti attivi per una determinata reazione catalitica.
Il problema di definire quali e quanti atomi concorrano a formare il sito e quanti centri attivi esistano per unità di superficie è di grande importanza per comprendere il ruolo della superficie stessa. Al problema si è cercato di dare una risposta con tecniche diverse, dall'impiego degli isotopi radioattivi, a tecniche di dosaggio di ‛veleni' che blocchino e titolino i siti. Nel caso dei metalli il problema del numero di atomi coinvolti è stato affrontato spesso con l'impiego di leghe formate da un metallo M attivo e da un metallo P non attivo. Per esempio, si abbia la molecola n-esano, CH3−(CH2)4−CH3, che può essere deidrogenata e ancorata su una superficie di atomi M. Se per arrivare alla rottura del legame C−C è necessario l'ancoraggio in due punti, i due atomi M dovranno essere vicini. Essi costituiscono l'‛insieme' (ensemble) attivo per la rottura (v. Sachtler e van Santen, 1977). Se in una lega gli atomi M sono diluiti in atomi P, essi saranno statisticamente lontani e il numero di insiemi sarà fortemente ridotto: sarà favorito un unico punto di attacco. Le conseguenze dal punto di vista chimico saranno importanti ai fini della successiva evoluzione: nel caso citato (atomi M diluiti) sarà meno favorita la deidrogenazione spinta e la rottura (cracking) della molecola. Oltre all'aspetto puramente geometrico, però, l'esistenza di atomi P vicini a M modificherà la struttura elettronica esistente su M, o esistente in un piccolo gruppo di atomi M circondati da P. Questi atomi che attorniano il sito possono essere indicati con il termine generico di ‛ligandi' o ‛leganti' in analogia alla chimica dei composti di coordinazione. In alcune trattazioni si mette in luce questo ‛effetto dei leganti' (ligand effect; ibid.). La metodologia spettroscopica ha permesso di dare importanti contributi all'impostazione del modello del sito.
3. La superficie come sede di siti a più funzioni.
a) Stati ionici superficiali.
Dal punto di vista geometrico è intuitivo che gli atomi o ioni superficiali si trovino in situazione intrinsecamente diversa da quelli interni. Il concetto di insaturazione dei legami è stato espresso fin da quando la diffrazione dei raggi X ha mostrato l'ordine esistente all'interno dei cristalli, ed è stato alla base del concetto di ‛centro attivo' formulato da H. S. Taylor (1925). La valutazione degli effetti di terminazione del cristallo in termini fisici è venuta in tempi successivi, con i progressi della fisica dei solidi.
Nell'articolo superfici: Fisica delle, suppl., si è visto che la perturbazione causata dall'esistenza di una superficie introduce livelli di energia permessi per gli elettroni, localizzati in prossimità della superficie, all'interno della ‛zona proibita' (forbidden gap) nel diagramma energetico (v. superfici: Fisica delle, suppl., fig. 18). Questi ‛stati superficiali' non dipendenti da impurezze o da difetti diversi dalla superficie stessa sono detti ‛intrinseci'. Da un punto di vista teorico è stata comunemente adottata una distinzione a seconda che gli stati intrinseci appartengano a solidi, quali i solidi covalenti semiconduttori elementari come il Si, in cui si abbia una grande sovrapposizione di orbitali e una ‛ibridazione', o a solidi, quali i solidi ionici, composti da più specie atomiche con differenti elettronegatività, e che risultano isolanti o semiconduttori con ampia zona proibita. Nel primo caso gli stati di superficie sono detti ‛stati di Shockley' e comportano l'esistenza di orbitali disponibili per legare gli atomi di superficie o tra loro, o con atomi esterni (dangling bonds). Nel secondo caso (solidi ionici) si parla di ‛stati di Tamm', o ‛stati ionici'. Dato l'interesse per le proprietà chimiche dei solidi ionici e la loro importanza in fenomeni di superficie, è utile descrivere in maggior dettaglio questi stati ionici e mostrare come si modifichi il carattere chimico degli atomi (ioni) di superficie.
Si prenda come esempio un composto MX, con X più elettronegativo, quale NaCì. Si esamini nella fig. 3 come varia l'energia E in funzione della distanza r di separazione tra M e X (v. Morrison, 1977). Lo zero dell'energia è preso con l'elettrone all'infinito e fermo e corrisponde allo stato X + M+ + e. A r = ∞ il livello di M sarà sotto lo zero della quantità IM (energia di ionizzazione di M), e quello di X della quantità AX (affinità elettronica di X). In genere il guadagno di energia per la reazione X + e = X- non è sufficiente a compensare la perdita per la reazione M = M+ + e, e, come nella figura, il livello di M si trova sotto quello di X. Se si diminuisce r, l'energia di attrazione coulombiana va aggiunta al bilancio energetico globale, e può aver luogo il trasferimento elettronico. In pratica, gli atomi X posti vicino a M diminuiscono il valore di IM, e il corrispondente livello sale, mentre gli atomi M aumentano il valore assoluto di AX, e il livello di X scende (curve continue). L'entità dell'interazione coulombiana dipende però dalla geometria della disposizione di M e X: all'interno del solido essa è aumentata di un fattore, detto ‛fattore di Madelung', rispetto a quella esistente in una molecola biatomica isolata (per es. in NaCl il fattore è 1,748). Gli atomi di superficie, non godendo della piena coordinazione, non potranno beneficiare dello stesso fattore di Madelung, e l'effetto sarà minore (curve tratteggiate). Pertanto, mentre i livelli di M e X appartenenti all'interno si troveranno nei punti C e V, una volta raggiunta la distanza di separazione, r0, esistente nel cristallo, i livelli pertinenti agli atomi superficiali si troveranno in C′ e V′, rispettivamente più basso e più alto di C e V. I punti C′ e V′ caratterizzano quindi livelli elettronici confinati alla superficie, detti appunto stati ionici superficiali o stati di Tamm. La separazione tra C′ e V′ è minore di quella tra C e V, cioè la ionicità in superficie è diminuita. Lo stato C′ agirà da ‛accettore' e quello V′ da ‛donatore' di elettroni. I cationi M+ e gli anioni X- superficiali avranno, rispettivamente, più affinità per l'elettrone e più tendenza a donarlo dei corrispondenti ioni situati all'interno del solido. In generale, la maggiore insaturazione diminuisce il carattere ionico del legame.
b) Struttura e proprietà di siti in solidi reali.
Il fatto che la superficie di un cristallo non sia una superficie geometrica, in cui tutti i punti godono delle stesse proprietà, fa sorgere la necessità di formulare modelli di superficie che tengano conto delle possibili situazioni a livello atomico e microscopico. Un cristallo può presentare facce diverse, terrazze, spigoli, ecc. (v. superfici: Fisica delle, suppl., fig. 5) e difetti puntiformi (lacune, interstiziali). I siti superficiali avranno quindi proprietà diverse a seconda della coordinazione. In un solido ionico M, per esempio un ossido ionico come MgO, deve essere tenuto presente il fatto che le particelle costituenti sono ioni. Si consideri la superficie di MgO, in particolare la faccia (100), che presenta cationi e anioni in numero uguale ed è quindi elettricamente neutra (v. fig. la): si troveranno esposti idealmente sia cationi Mg2+ sia anioni O2-. Una molecola polare, come HCl o H2O, sarà attratta dalla superficie in modo differenziato da punto a punto, in quanto la parte positiva della molecola polare interagirà con gli ioni negativi della superficie e la parte negativa con gli ioni positivi. Si vedrà, nel È c, che ciò configura l'esistenza di siti con proprietà diverse. Si consideri ora un piano non elettricamente neutro, per es. (111), composto solo da cationi o da anioni (v. fig. 1c). Dato che il cristallo è nel suo insieme elettricamente neutro, esso dovrà terminare con un numero uguale di facce positive e negative. Tale situazione non è energeticamente favorevole, e infatti si avrebbe una diminuzione di energia spostando parte degli anioni di una faccia sui cationi dell'altra faccia, scoprendo così in parte il piano sottostante. La ‛superficie' così creata presenta sia cationi che anioni, ma su piani diversi. Come ultimo esempio di superfici che intrinsecamente presentano situazioni geometriche e quindi siti particolari si può citare la faccia (110), dove sono presenti filari di cationi e di anioni (v. fig. lb). Un esempio verrà illustrato in seguito nel caso dell'allumina γ-Al2O3.
Si è già notato che la superficie di un cristallo si trova in una situazione di particolare reattività. Gli esempi sopra riportati si riferiscono quindi solo a condizioni ideali nel vuoto. Normalmente, tranne i casi particolari di vuoti molto spinti, la superficie è esposta a un ambiente: un caso comune è l'esposizione a un gas contenente acqua. Si può vedere la conseguenza di questa interazione considerando, per esempio, una faccia (100) di MgO. Se per semplicità si omettono le cariche ioniche, la superficie e la sua reazione con H2O possono essere schematizzate come segue:
La superficie terminerà quindi con gruppi idrossile OH, con diminuzione dell'energia superficiale e stabilizzazione della struttura. La disidratazione delle superfici così composte porterà alla formazione di ioni coordinativamente non saturi, detti ioni o siti CUS (Coordinatively Unsaturated Surface ions o sites). I siti CUS hanno reattività particolare e pertanto il processo di formazione e i trattamenti subiti dal solido determineranno la chimica della superficie, non descrivibile dalla sola formula o composizione media.
È infine da citare, senza entrare in particolari, il caso dei solidi finemente dispersi, nei quali una frazione elevata di atomi si trova in superficie. Viene definito ‛dispersione' D il rapporto tra atomi di superficie e atomi totali. Se D ha valori elevati, prossimi al valore massimo 1, la maggioranza degli atomi è esposta e tutto il solido può presentare caratteristiche differenti dal composto massivo. I solidi dispersi hanno notevole importanza applicativa - come catalizzatori e come adsorbenti - e teorica: specie nel caso dei metalli e delle leghe, si stanno compiendo studi di carattere teorico e ricerche sperimentali concernenti diverse proprietà fisiche (magnetismo, energia di estrazione, proprietà ottiche, ecc.) dei solidi dispersi.
c) Proprietà acide e basiche.
I concetti di acido e di base, inizialmente introdotti per la chimica delle soluzioni acquose, hanno subito un'evoluzione che ne ha permesso l'estensione a situazioni in cui non è presente l'acqua, e quindi anche a reazioni che coinvolgono la superficie di un solido. Le due accezioni più comunemente impiegate in chimica sono quella di J. N. Brönsted (B.), secondo cui un acido è un donatore di protoni e una base è un accettore di protoni, e quella di G. N. Lewis (L.), secondo cui un acido è un accettore di una coppia elettronica e una base è un donatore di una coppia elettronica. In molti casi una superficie mostra siti che hanno caratteristiche di acidi di B. o di L., o di basi di B. o di L. Alcuni esempi chiariranno i concetti e permetteranno un esame di alcune superfici tipiche. Lo ione O2- è in linea di principio sia un accettore di protoni H+ (base di B.) sia un donatore di coppia elettronica (base di L.). Il gruppo OH- esistente su superfici di ossidi idratati o di idrossidi può agire da base di B. (acquistando H+) o da acido di B. (dissociandosi in O2- + H+). Uno ione metallico quale Al3+ è un accettore di coppia elettronica, presentando in superficie degli orbitali vuoti, e quindi è un acido di L. Quindi ioni tipici presenti in superficie hanno in linea di principio una varietà di caratteristiche acide e basiche. Inoltre le funzioni acide e basiche possono coesistere sulla stessa superficie in siti diversi.
Si è parlato finora in modo qualitativo dell'acidità secondo B. o secondo L., ma è logico che, come nelle soluzioni, interessa conoscere sia la ‛quantità' (concentrazione) sia la ‛qualità' (forza acida) dei siti attivi. Vi sono modi diversi per affrontare il problema, e si può dire che, a seconda del sistema, può essere realizzabile e consigliabile un metodo piuttosto che altri. Per esempio, si può misurare mediante termogravimetria la quantità di una base gassosa legata dalla superficie e poi la perdita di base in funzione della temperatura, ottenendo così una misura di concentrazione e una distribuzione della forza dei siti. L'applicazione della spettroscopia IR verrà ricordata nel cap. 5, È a. Infine un metodo spesso impiegato, analogo concettualmente a quello usato nel caso delle soluzioni, è il metodo degli indicatori.
Anche per le superfici di solidi può essere definita la forza degli acidi (e delle basi) superficiali, secondo i principi applicati alle soluzioni. Se si considera l'equilibrio
BH+ ???01??? H+ + B
e la costante di equilibrio corrispondente
Ka = aH+aB/aBH+ = aH+ (fBcB/fBH+cBH+),
dove a sono le attività e f i coefficienti di attività delle singole specie, si può passare ai logaritmi e, ricordando la definizione pK = − log K, ottenere l'equazione generale
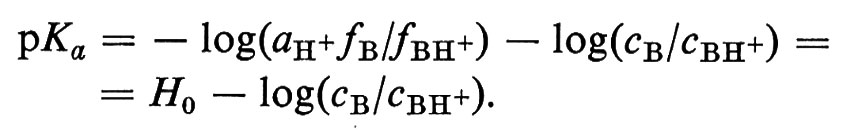
La funzione H0, detta funzione di Hammett, misura la forza acida: in soluzione acquosa diluita essa tende al valore di pH. Mediante una serie di indicatori è possibile avere una misura del valore di H0 e definire la distribuzione della forza dei siti acidi esistenti sulla superficie di un solido. Alcuni solidi hanno siti dotati di forza acida molto notevole (valori di H0 molto negativi), equivalente a quella di un acido forte e concentrato. Essi presentano proprietà dette di superacido, di notevole interesse in reazioni catalitiche. Esempi di superfici che presentano siti acidi forti sono le silico-allumine attivate (v. È d), mentre basica è la superficie di MgO. La γ-Al2O3 mostra la presenza di siti acidi dopo riscaldamento (v. È d).
d) Allumina γ e zeoliti: due casi tipici nella chimica delle superfici.
Allumina. - La γ-Al2O3 è una delle forme in cui si presenta l'ossido di alluminio e fa parte delle cosiddette allumine di transizione, che ad alta temperatura si trasformano nella forma α-Al2O3 (corindone). La γ-Al2O3 ha struttura cubica tipo spinello, ma difettiva, in quanto non tutte le posizioni cationiche sono occupate. Nello spinello esistono posizioni cationiche a coordinazione tetraedrica (t) e ottaedrica (o). I piani reticolari paralleli alle facce (111) della struttura ideale sono di due tipi, A e B, e sono rappresentati nella fig. 4. In A si hanno cationi in siti tetraedrici e ottaedrici, mentre in B i cationi sono solo in posizione ottaedrica. I piani tipo (110) sono pure di due tipi, C e D, rappresentati anch'essi nella fig. 4: si noti la presenza di filari di cationi, ottaedrici e tetraedrici in C, solo ottaedrici in D.
Anche nella γ-allumina la terminazione dei cristalliti avviene con idrossili OH e non con ioni ossigeno. La struttura permette però di distinguere diversi tipi di OH a causa delle possibili configurazioni, come riportato nello schema per tre di esse (Ia, legato ad Al t; IIa, a ponte tra o e t; IIb, a ponte tra o e o).
In totale sono distinguibili 5 configurazioni, che secondo Knözinger e Ratnasamy (v., 1978) giustificano la comparsa di 5 bande distinte OH nell'infrarosso.
Quando si disidrata l'allumina, la perdita di acqua non avviene in maniera uniforme, ma alcune configurazioni tendono a rimanere sulla superficie (ibid.). Ciò ha portato a una distinzione nel tipo di acidità e nell'interazione con molecole diverse, e quindi a una chimica di superficie dovuta alla coesistenza di siti con caratteristiche proprie.
Zeoliti. Esistono diverse classi di zeoliti, la cui struttura è stata determinata con precisione e che mostrano delle cavità interne connesse da canali (v. Breck, 1974; v. Gates e altri, 1979). L'unità chimica di partenza è un tetraedro di ioni O2- uniti a uno ione Si4+ (v. fig. 5A). Alcuni ioni Si4+ possono essere sostituiti da Al3+; data la minore carica positiva di questo ione, la neutralità elettrica sarà mantenuta con cationi addizionali esterni, quali Na+. Queste unità chimiche si uniscono tramite gli ossigeni in una struttura base della sodalite, che è un ottaedro tronco (v. fig. 5B). Le unità strutturali sodalitiche possono essere unite tramite ponti ossigeno o tra facce a 4 membri (v. fig. 6A), come nella zeolite A, o tra facce a 6 membri, come nella faujasite o nelle zeoliti X e Y (v. fig. 6B). È importante notare che nella struttura finale della zeolite vi sono due tipi di cavità, una grande (al centro della fig. 6B), detta α, e una piccola entro la ‛gabbia' sodalitica, detta β. La terza cavità γ, nei prismi, è troppo piccola per esplicare una funzione di facile scambio con l'esterno, cioè di ‛superficie'. Corrispondentemente si hanno differenti posizioni cationiche, visibili nella sezione della fig. 7. I cationi Na+ che bilanciano la carica negativa possono occupare le diverse posizioni e possono essere scambiati con altri cationi, in rapporto tale da bilanciare sempre la carica totale. Si può rappresentare la chimica che ha luogo sulle superfici interne secondo i seguenti schemi:
A temperatura moderata (circa 350 °C) lo ione NH4+ si decompone liberando NH3 e lasciando il protone, che verrà legato a un ossigeno (c). Si forma quindi un acido di Brönsted:
A temperatura più elevata (circa 550 °C) avviene una disidratazione, che lascia Al- (donatore di coppia elettronica, cioè base di Lewis) e ioni Si+ scoperti (accettori di coppia, quindi acidi di L.) (d).
La chimica di superficie delle silico-allumine e delle zeoliti può essere resa ancora più complessa mediante l'introduzione di ioni bivalenti (per es. Mg2+) o trivalenti, in particolare lantanio e lantanidi, noti come ‛terre rare' e quindi spesso indicati con la sigla RE (Rare Earths). Per semplicità la superficie degli schemi da (a) a (d) viene simboleggiata dai soli gruppi Al (e Al-), e si avrà allora
3Al-Na+ + RE3+ = (Al-)3RE3+ (e)
e a seguito di idrolisi parziale
(Al-)3RE3+ + H2O → (Al-)2RE3+(OH-) + Al OH. (f)
In (f), come si può notare dagli schemi più estesi precedenti, l'idrossile non è posto solo vicino all'Al, ma anche al Si. L'OH si legherà come in (e) al Si, dando luogo a un'acidità di B. Per riscaldamento a più alte temperature avviene una disidratazione e, come in (d), si manifesta acidità secondo L. La fig. 8 mostra la variazione della concentrazione dei siti di B. e di L. con la temperatura, misurata mediante spettroscopia IR (v. cap. 5, È a) in una zeolite Y contenente Mg.
e) Chimica dei clusters e chimica delle superfici.
In relazione ai problemi discussi sulla definizione del sito (numero di atomi partecipanti, effetto dei leganti, struttura elettronica) è da citare un settore della chimica dei composti di coordinazione che apre promettenti prospettive anche per la chimica delle superfici: la chimica dei clusters. La parola inglese cluster indica un piccolo insieme di oggetti; essa è entrata nel linguaggio scientifico con significati specifici alquanto diversi, sempre legati al concetto di ‛piccolo insieme' o ‛piccolo gruppo'. Nella chimica dei composti di coordinazione un cluster è un composto molecolare formato da un certo numero di atomi metallici legati tra loro e stabilizzati da leganti L. La struttura di molti clusters è stata determinata con precisione mediante diffrazione di raggi X, e sono state studiate molte proprietà dei legami mediante tecniche spettroscopiche. Appartengono a questa famiglia i carbonili polinucleari, come per esempio il composto Ir4(CO)12 . La sostituzione parziale di CO con trifenilfosfina facilita la dissociazione dei rimanenti CO, con un effetto simile, in linea di principio, a quello citato (v. cap. 2, È b) nella descrizione degli ‛insiemi' e dell'effetto dei leganti. Altri esempi, di notevole complessità, sono l'anione trimero [Pt3(CO)3(μ2−CO)3]32- (v. fig. 9A), e l'anione [Rh13(CO)25H5-n]n- (v. fig. 9B). Si noti nel primo l'embrione di una struttura metallica a strati, e nel secondo il nucleo di atomi metallici disposti come nella struttura esagonale compatta.
I clusters possono essere visti come ponte intermedio tra strutture molecolari e piccoli cristalli metallici, e rappresentano un interessante modello per discutere i problemi sopracitati. Nel cluster possono essere contenuti atomi metallici diversi, come nelle leghe, e ciò rappresenta un ulteriore motivo di interesse.
La parola cluster designa anche un piccolo insieme di atomi metallici, che possono essere ottenuti per dispersione su un supporto costituito da un ossido inerte o da un polimero. Affinché questi clusters non sinterizzino è necessaria un'interazione stabilizzante con il supporto, i cui atomi svolgono quindi una funzione di legante. I clusters metallici possono essere costituiti da atomi diversi. Si comprende come non esista un confine netto tra il primo e il secondo significato, e la continuità è ancora maggiore se si considera il caso, oggi molto studiato, di clusters molecolari supportati. Esiste in questo caso il problema di determinare la struttura dopo l'interazione e di verificare quindi l'integrità della molecola originale o il tipo di trasformazione, problema oggi in genere affrontato con tecniche spettroscopiche, tra le quali IR, PES, EXAFS.
4. Termodinamica delle superfici e composizione superficiale.
La termodinamica ha rivolto la sua attenzione alle superfici sin dall'inizio del suo sviluppo, oltre 100 anni or sono, principalmente ad opera di J. W. Gibbs (1875), nella trattazione delle soluzioni liquide. Già allora fu messo in evidenza che la composizione dello strato superficiale del liquido poteva differire da quella dell'interno. Gli stessi principi termodinamici governano l'interfaccia di un solido, sia per quanto riguarda l'arricchimento di una componente gassosa alla superficie solida (adsorbimento), sia per quel che concerne la composizione della superficie del solido. Il fenomeno dell'adsorbimento di gas su solidi indusse I. Langmuir (1918) a formulare un modello di gas ‛bidimensionale', dal quale si svilupparono poi modelli più elaborati (oggi direttamente visibili con la diffrazione di elettroni lenti, LEED), che includevano la possibilità di ordine, di transizioni di fase, ecc. Data l'importanza della termodinamica dei processi di adsorbimento in problemi di cinetica dei reattori, in catalisi eterogenea e nelle separazioni di fasi (cromatografia analitica e preparativa), questi problemi sono in genere trattati nei rispettivi settori (v. Cimino e Carrà, 1980; v. Flood, 1967; v. Clark, 1970). Come ricordato nel cap. 1, nella chimica delle superfici si tende oggi a favorire gli aspetti microscopici dei fenomeni di superficie, e quindi la termodinamica dei processi di adsorbimento non verrà qui trattata. È però di interesse specifico per l'interazione molecola-superficie il problema della composizione superficiale di un solido. In questo campo si è andata sviluppando una correlazione tra le grandezze macroscopiche impiegate in termodinamica e quelle microscopiche chimiche (natura del legame chimico, ruolo delle impurezze, ecc.), man mano che nuove tecniche hanno permesso un'indagine più accurata e quantitativa dei fenomeni di superficie. Per esempio, la termodinamica dimostra che la creazione di una superficie richiede energia e che pertanto un solido tende ad avere il minimo sviluppo superficiale (v. Swalin, 19722). Esiste quindi un'‛energia superficiale' σ (dimensioni: energia/superficie = forza/lunghezza) che rappresenta l'energia richiesta per creare un'unità di superficie. La grandezza σ (positiva) è tanto più grande quanto più difficile è l'incremento di superficie, quindi quanto più difficile è rompere i legami esistenti in un solido per poterne estendere la superficie. È quindi logico attendersi una correlazione tra σ ed entalpia di sublimazione ΔHsub, essendo quest'ultima una misura della forza di legame. Un esempio di questa correlazione è illustrato nella fig. 10, che riporta l'energia superficiale molare, σm = N1/3 V2/3 σ (N = numero di Avogadro, V = volume molare) in funzione di ΔHsub.
Dal punto di vista sperimentale non è agevole la misura di σ, essendo questa fortemente influenzata da impurezze provenienti o dal materiale stesso o dall'ambiente.
Un'importante conseguenza a livello microscopico, e quindi sul comportamento chimico di una superficie, è il fatto che un solido costituito da due specie A e B, anche se perfettamente omogeneo e con dispersione atomica all'interno, non mostrerà in genere la stessa composizione in superficie. La trattazione termodinamica risulta più o meno complessa a seconda che si considerino soluzioni reali o ideali. Per queste ultime si assume che non esista una preferenza nella formazione dei legami A−A, B−B, o A−B, cioè che l'entalpia di mescolamento ΔHm sia nulla. In questa ipotesi si può dimostrare che la composizione superficiale, descrivibile in termini pratici dal rapporto tra le frazioni molari x di A e di B esistenti in superficie, è data dalla relazione (v. Sachtler e van Santen, 1977; v. Swalin, 19722; v. Defay e altri, 1966; v. Somorjai, 1981)
x Bs /x As = (x Bb /x Ab) exp(a Δσ/RT),
dove l'indice b si riferisce all'interno del solido, s alla superficie, a è l'area molare di A e di B (per semplicità assunte uguali) e Δσ = σA − σB è la differenza di energie superficiali tra A e B puri.
La relazione mostra che la superficie si arricchisce del componente che abbassa l'energia superficiale. Una trattazione più generale prende in esame non solo l'energia superficiale, ma anche la differenza di dimensioni tra atomi (o ioni, in un composto ionico) A e B, differenza che tenderà a introdurre deformazioni che possono essere minimizzate arricchendo la superficie con il componente avente dimensioni maggiori. Infine, il caso delle soluzioni reali introduce un ulteriore parametro da prendere in esame: il valore di ΔHm (≠ 0) (v. Somorjai, 1981; v. Wynblatt e Ku, 1977; v. Sachtler, 1984). Il problema della composizione superficiale costituisce un campo di indagine vivo e in notevole sviluppo, che si avvale di molteplici tecniche (v. cap. 5), in particolare delle spettroscopie elettroniche Auger e XPS (X-Ray Photoelectron Spectroscopy) e dello scattering ionico, ISS. Si comprende comunque che la reattività della superficie risente della composizione, con importanti conseguenze in svariati fenomeni di interesse in settori applicativi (corrosione, catalisi eterogenea, microelettronica).
Sono ormai disponibili esempi di arricchimento superficiale di numerose leghe metalliche binarie, studiate con tecniche differenti. La fig. 11 illustra il caso del sistema Pt−Cu, che mostra l'accordo tra le varie tecniche e l'accordo soddisfacente con la teoria che tiene conto della non idealità della soluzione (effetti di deformazione dovuti alle dimensioni atomiche) (v. Sachtler, 1984). Anche nel caso di soluzioni solide di ossidi è possibile lo studio della composizione superficiale (v. Cimino, 1985).
Dal punto di vista chimico è interessante anche il caso di leghe bifasiche, in quanto possono verificarsi diverse morfologie che comportano superfici esposte arricchite in un particolare componente. Non sempre, infatti, le due fasi si presentano come domini distinti e ugualmente esposti. A volte una fase tende a coprire l'altra, con una morfologia detta ‛a ciliegia' (cherry model), in quanto vi è uno strato esterno come la polpa nel frutto. Come esempio può essere citato il caso classico del sistema rame-nichel, un sistema che solo in tempi recenti si è potuto investigare a fondo.
Il sistema Cu−Ni mostra una lacuna di miscibilità al di sotto di circa 320 °C. Esiste una tendenza all'arricchimento superficiale in Cu, messa in evidenza mediante tecniche fisiche spettroscopiche. In dipendenza dalla composizione si possono distinguere vari intervalli schematicamente rappresentati nella fig. 12 (v. Sachtler e van Santen, 1977). Designata con x la frazione molare del Cu e con x1 e x2 i valori critici che definiscono la lacuna, si ha una prima zona monofasica, per x2 〈 x 〈 1, in cui si ha arricchimento in Cu (regione 1). Per x1 〈 x 〈 x2 si hanno due fasi, una esterna ricca in Cu e una interna ricca in Ni (regione 2).
Un caso particolare è dato da x1 〈 x 〈 (x1 + Δx), dove la concentrazione del Cu non è sufficiente alla copertura del nucleo interno ricco in Ni, che viene così parzialmente esposto. Infine, se 0 〈 x 〈 x1 (regione 3), si ha un'unica fase ricca in Ni. Si può capire che la chimica di superficie sarà fortemente dipendente dall'effettiva composizione dei piani esterni. Per esempio, nel caso 2 una diminuzione di concentrazione x non si riflette in modo proporzionale nella composizione esterna (e infatti si assottiglia lo strato esterno) e quindi vi è apparente indipendenza delle proprietà dalla composizione globale. Ciò si riflette, per esempio, nelle proprietà di adsorbimento di idrogeno (v. fig. 13; v. Sachtler e van Santen, 1977).
5. Esempi di applicazioni di tecniche spettroscopiche a problemi di superficie.
La tendenza della chimica delle superfici a procedere per problemi, già citata nel cap. 1, e le potenzialità offerte dalle nuove tecniche messe a punto dalla fisica delle superfici hanno caratterizzato il corso degli ultimi 30 anni. Il salto di qualità è venuto dallo sviluppo delle tecniche spettroscopiche, che hanno affiancato le metodologie tradizionali della chimica, peraltro spesso elaborate sperimentalmente e teoricamente per essere adattate a problemi di superficie. Il processo di sviluppo delle tecniche spettroscopiche è ancora in pieno svolgimento, ed è da attendersi che continuerà a portare risultati importanti anche nel futuro prossimo. Nell'articolo superfici: Fisica delle, suppl., sono stati illustrati i principi fisici sui quali si basano le principali tecniche di superficie, che forniscono un gran numero di informazioni spesso complementari. La tab. I riporta un elenco delle principali tecniche impiegate nella caratterizzazione di superfici, il loro acronimo (dalle iniziali dei nomi in inglese) e il tipo di informazione fornita. Alcune non sono specifiche per sole proprietà di superficie (per es. ESR, NMR), ma sono state utilmente impiegate nella chimica e nella fisica delle superfici. Altre tecniche di superficie non elencate hanno trovato applicazione più limitata o sono in corso di sviluppo. Si comprende l'impossibilità di trattare, nei limiti di un articolo, le numerose applicazioni di queste tecniche nè, d'altra parte, un semplice elenco di applicazioni può aiutare a comprendere la ricchezza di risultati raggiunti e le prospettive aperte. Si preferisce quindi dare alcuni esempi di applicazioni di tre tecniche (IR, PES e ESR) a problemi particolari. Per altri esempi si vedano Morrison, 1977, cap. 3, e Somorjai, 1981, cap. 2.
a) Spettroscopia infrarossa.
È stato già ricordato nel cap. 2 che le specie molecolari presenti sulla superficie sono simili a quelle che si trovano libere nei gas o disperse omogeneamente in soluzione. In linea di principio è quindi possibile applicare le stesse tecniche volte alla definizione della struttura molecolare impiegate in chimica. Tra le tecniche, le spettroscopie che studiano lo spettro vibrazionale delle molecole sono di importanza fondamentale, poiché permettono di studiare direttamente le proprietà del legame. Lo studio degli spettri di vibrazione può essere realizzato con tecniche diverse: spettroscopia nell'infrarosso (IR), spettroscopia Raman, spettroscopia per perdita di energia elettronica (EELS). Per l'ampiezza delle sue applicazioni, ormai acquisite da circa 30 anni, e per l'importanza che tuttora ha, la tecnica spettroscopica IR è di particolare rilievo (v. Delgass e altri, 1979). La prima applicazione alla chimica delle superfici, effettuata in relazione a problemi di catalisi eterogenea e realizzata mediante misure in trasmissione, risale a circa la metà degli anni cinquanta. Il metodo impiegato implicava il superamento di una grossa difficoltà; infatti gli atomi superficiali di un solido sono dell'ordine di 1015 per cm2, con uno spessore di ogni strato atomico dell'ordine di 2 × 10-8 cm. La luce penetra per una profondità di circa 10-5 o 10-6 cm e quindi coinvolge da 50 a 500 strati del solido. Anche se gli atomi o le molecole adsorbiti sono in numero paragonabile al numero degli atomi esterni, si è di fronte al problema di dover studiare una minoranza dei possibili contributi allo spettro. Si può con vari accorgimenti massimizzare il contributo delle specie di superficie, ricorrendo a solidi il più possibile trasparenti, a campioni policristallini che presentino un elevato sviluppo superficiale e quindi un'elevata concentrazione di siti per unità di volume, e infine studiando molecole adsorbite che abbiano un elevato coefficiente di estinzione, e quindi siano visibili anche in concentrazione limitata. In altri casi (cristalli singoli), in cui è impossibile realizzare lo sviluppo superficiale elevato, proprio perché interessati a una specifica faccia cristallina, si è ricorsi a tecniche di riflessione. Infine la spettroscopia Raman ha fornito indicazioni simili ma complementari, spesso eliminando ambiguità nei risultati. In tempi più recenti si è aggiunta la tecnica EELS ed è prevedibile un'estensione di questa spettroscopia (v. Willis e altri, 1983).
In questo paragrafo si citeranno alcuni esempi con lo scopo di mostrare, da un lato, alcuni aspetti della chimica delle superfici e, dall'altro, l'applicazione della spettroscopia IR. In particolare si mostreranno: 1) l'effettivo parallelismo esistente in linea di massima tra molecole libere e specie legate alla superficie; 2) esempi di identificazione di specie superficiali; 3) e 4) la distinguibilità dei siti e delle loro proprietà mediante l'esame IR di specie a essi legate; 5) la reattività di specie superficiali e la formazione di possibili precursori nelle reazioni catalitiche.
1. Uno dei primi casi studiati è stato l'adsorbimento di CO, in cui poteva essere notata la comparsa di bande di assorbimento IR attribuibili a carbonili superficiali. Poteva essere distinta la formazione di carbonili lineari, M−CO, e di carbonili a ponte,
dove M è l'atomo metallico appartenente alla superficie. Lo studio della chimica inorganica forniva un'ampia base per discutere il tipo di legame: per esempio, potevano essere distinti legami σ e legami σ−π, questi ultimi con frequenze più basse (retrodonazione di elettroni, o back-donation). Molte specie di superfici presentano strette analogie con specie coordinate a un atomo centrale, quali si trovano in composti di coordinazione studiati in fase omogenea. La fig. 14 mostra una carta di confronto dove sono stati riportati gli intervalli in cui si osservano le frequenze nei composti di superficie (rettangoli bianchi) e quelli presenti in complessi omogenei (rettangoli neri). È chiaro che il confronto tra specie omogenee e adsorbite non solo è utile per l'attribuzione della struttura molecolare della specie di superficie, sulla base della molecola omogena, ma costituisce esso stesso un punto di partenza per lo studio delle differenze esistenti tra complessi omogenei e complessi di superficie.
2. La teoria dell'assorbimento IR definisce la dipendenza dalle masse dei numeri d'onda ν̄ (= 1/λ, con λ lunghezza d'onda). Pertanto una via utile per l'attribuzione delle bande spettroscopiche fa ricorso all'esame di spettri ottenuti con molecole isotopiche.
Per adsorbimento di idrogeno su ZnO si forma un idruro superficiale; si può ipotizzare che il processo sia
con formazione quindi di due legami, Zn−H e O−H. Mediante IR si identificano sia la banda idrossilica OH, sia la banda idrurica Zn−H. Per attribuire con maggiore certezza quest'ultima si sostituisce l'isotopo deuterio all'idrogeno. Si osservano così nuove bande; la tab. II riporta i numeri d'onda osservati (in cm-1). Se il solido fosse rigido, il rapporto ν̄H/ν̄D varrebbe √-2 = 1,414; se viceversa gli atomi Zn o O, a cui ciascun atomo H (o D) è legato, fossero liberi, i calcoli fornirebbero i valori riportati nella tabella, che sono in buon accordo con i dati sperimentali. L'accordo migliorerebbe se si considerasse che lo Zn e l'O non sono realmente liberi, ma accoppiati alla struttura del cristallo.

Lo studio di molecole contenenti isotopi è utile non solo per l'effetto dovuto alla massa, ma anche in quanto permette di seguire dove avviene la rottura del legame. Un classico esempio è la reazione tra propene CH3−CH=CH2 e ZnO. Si nota la formazione di un gruppo OH, e l'impiego di molecole marcate con deuterio mostra che si è distaccato l'H del gruppo CH3 (idrogeno allilico), con formazione di un complesso π-allile con la superficie.
3. A proposito delle superfici interne nelle cavità delle zeoliti, si è detto che esistono posizioni caratterizzate da diversa accessibilità. La tecnica IR ha molto contribuito allo studio delle proprietà di superficie (interna) delle zeoliti e ha tra l'altro permesso di mettere in evidenza la differente accessibilità dei siti. Nelle zeoliti tipo Y, ottenute per scambio con NH4+ e decomposizione a 350 °C, si notano due tipi di gruppi OH con ν̄ = 3.650 e 3.550 cm-1, entrambi con carattere acido, dato che le bande sono eliminate per reazioni con NH3. Se si usa una base di dimensioni ingombranti, quale la piridina C6H5N, solo la banda a 3.650 cm-1 scompare. Si è potuto dedurre che l'OH che non reagisce con la piridina deve trovarsi nelle cavità più piccole, β, della struttura zeolitica (v. fig. 7).
4. La spettroscopia IR ha permesso di distinguere tra siti acidi di B. e di L. Per esempio, adsorbendo piridina, nel caso dei siti acidi di B. si forma lo ione piridinio, che presenta bande distinte da quelle della piridina coordinata a un sito acido di L. La possibilità di distinguere i due siti ha permesso di studiare non solo la variazione delle rispettive concentrazioni con trattamenti diversi, quale il semplice riscaldamento già citato in precedenza (v. fig. 8), ma anche il ruolo di ciascuna specie in reazioni catalitiche.
5. Un aspetto che è andato assumendo importanza crescente, e che ha ancora un notevole potenziale per il futuro, è lo studio delle reattività delle specie superficiali. Un esempio che riguarda un problema attuale è lo studio della reazione tra CO e idrogeno, base per l'utilizzazione di fonti di energia alternative al petrolio. Tra i catalizzatori impiegati per la sintesi di metanolo, CH3OH, vi sono quelli a base di ZnO. Se, dopo aver adsorbito idrogeno e sviluppato così la banda caratteristica Zn−H, si adsorbe CO a pressioni crescenti, si osserva un'evoluzione della banda Zn−H inizialmente presente, e designata I, con formazione di due nuove bande, II e III, dovute alla formazione di un complesso superficiale tra H e CO, precursore del prodotto di idrogenazione finale (v. fig. 15; v. Boccuzzi e altri, 1978).
b) Spettroscopia fotoelettronica.
La spettroscopia elettronica per fotoemissione, sia quella eccitata da raggi X (XPS o ESCA), sia quella eccitata da radiazione ultravioletta (UPS), è oggi largamente impiegata nel campo della chimica delle superfici e nelle problematiche a essa collegate (v. Briggs e Seah, 1983).
La tecnica consente sostanzialmente di misurare (v. cap. 4) tre grandezze: a) l'energia dei fotoelettroni; b) l'intensità della corrente elettronica registrata a ciascuna energia ('picco' fotoelettronico); c) la forma del picco.
L'energia dei fotoelettroni dà indicazioni sulla specie chimica, sul suo stato di ossidazione (per es. distingue Ni da Ni2+) e in casi favorevoli, e con opportuni accorgimenti strumentali che aumentano la precisione delle misure, dà anche indicazioni sull'intorno (per es. O di un ossido o di un idrossido).
L'intensità della corrente elettronica consente un'analisi quantitativa della superficie, fino a una profondità che dipende dall'energia (v. superfici: Fisica delle, suppl., fig. 7). Si ricordi infatti che la tecnica raccoglie gli elettroni che provengono da una profondità dell'ordine di 5-30 Å (0,53 nm), cioè da 2 a 15 strati atomici. La tecnica quindi non dà informazioni relative al solo ultimo strato; vi sono però diversi casi in cui ciò non costituisce una limitazione, ma addirittura offre un vantaggio. Per esempio, se vi è una data specie solo sullo strato esterno e i segnali relativi a questa specie non rischiano di confondersi con i segnali di specie differenti provenienti dall'interno, la tecnica in questione può avere notevole sensibilità, anche se in genere la spettroscopia Auger risulta più sensibile.
Tra le diverse indicazioni ricavabili dalla forma del picco le più frequentemente utilizzate nella chimica delle superfici sono quelle sulla presenza contemporanea di più specie. Le moderne tecniche di elaborazione dei dati consentono la deconvoluzione del picco, se non risolto, e la valutazione quantitativa dei singoli contributi.
È stato già detto che gli elettroni raccolti non provengono solo dall'ultimo strato: in molti problemi devono essere esaminati campioni policristallini contenenti impurezze superficiali e la ‛capacità' dei fotoelettroni di ‛vedere' sotto lo strato di impurezze consente lo studio del materiale. Dal punto di vista chimico particolarmente interessante è il confronto della superficie prima e dopo l'esposizione a un ambiente reattivo (corrosione, catalisi). Anche in questo caso l'inevitabile contaminazione con prodotti di reazione o con strati carboniosi non impedisce lo studio dei processi di superficie.
Un ulteriore vantaggio della tecnica fotoelettronica è la possibilità di studiare sia campioni policristallini, quali quelli impiegati in molti problemi applicativi, sia monocristalli o film, dalla composizione o struttura ben determinate. È così possibile estendere lo studio da materiali tipici della fisica delle superfici a quelli solitamente impiegati in chimica. Un esame di alcuni problemi particolari illustrerà l'applicazione della tecnica.
Ossidazione di metalli. - Si sono fatti notevoli progressi nella comprensione del fenomeno dell'ossidazione dei metalli, problema che interessa molti campi (corrosione, catalisi, elettrochimica). Grazie alle tecniche fotoelettroniche e alla spettroscopia Auger, si è potuta chiarire l'influenza di impurezze, la formazione iniziale di una specie ‛ossigeno' legata alla superficie, con caratteristiche diverse dall' ossigeno ossidico, la successiva penetrazione dell'ossigeno e la formazione di ossido. I risultati completano quelli di tipo strutturale (LEED-SIMS) ed elettronico (energia di estrazione). Si sono potute studiare le diverse facce di un cristallo e chiarire il ruolo della geometria superficiale. In campo applicativo si è studiato il fenomeno della formazione di ossidi protettivi e di precursori superficiali atti a favorire (o a ostacolare) l'adesione di rivestimenti protettivi (v. Briggs e Seah, 1983).
Adsorbimento di CO. - L'adsorbimento dell'ossido di carbonio, CO, è il primo passo di una serie di reazioni che includono la reazione tra CO e H2, di interesse nella sintesi di idrocarburi (sintesi di Fischer-Tropsch) e di alcoli (v. Biloen e Sachtler, 1981). Il problema della natura dell'interazione in dipendenza dal metallo ha ricevuto un decisivo chiarimento dalla spettroscopia elettronica. La molecola CO presenta spettri molecolari, visibili con UPS, distinti dagli spettri registrati dopo dissociazione in C + O. Inoltre anche il C dissociato può essere distinto da quello legato in CO. Si ottiene così una tabella di comportamento dei diversi metalli (v. Rhodin e Gadzuk, 1979; v. fig. 16) a seconda della loro posizione nel sistema periodico. La linea di demarcazione continua (v. fig. 16) separa i metalli che dissociano il CO a temperatura ambiente (a sinistra) da quelli che ne mantengono la forma molecolare (a destra); a temperatura più elevata la linea si sposta verso destra. Il Fe si trova al limite, con formazione lenta di C + O. La linea tratteggiata dà la demarcazione per l'adsorbimento dissociativo della molecola NO. La molecola N2 si comporta in maniera simile al CO. Questi studi hanno chiarito il ruolo di alcuni metalli, atti a formare alcoli e quindi a mantenere un legame C−O, rispetto ad altri dai quali si formano esclusivamente o prevalentemente idrocarburi.
Interazione di metalli con supporti e sistemi multicomponenti in catalisi. - Nella maggioranza delle applicazioni catalitiche delle proprietà di metalli è necessario disperdere il metallo attivo su un supporto inerte (Al2O3, SiO2, ecc.). L'effetto del supporto non è solo quello di disperdere la fase attiva e quindi di aumentare la superficie utile, ma anche quello di stabilizzare specie particolari. La tecnica XPS è stata particolarmente utile in questo campo, poiché ha molto contribuito a mettere in luce l'esistenza di un'interazione: per esempio, essa ha mostrato la resistenza alla riduzione di specie ossidiche disperse, dovuta all'interazione con il supporto (si sono potuti confrontare supporti diversi). Infine nei sistemi multicomponenti si è potuta mettere in evidenza la composizione superficiale reale (v. Barr, 1983).
Influenza del preadsorbimento di una specie A sulla reattività di una specie B. - Se si adsorbe acqua su una superficie pulita (111) di un monocristallo di Cu a circa 80 K (v. Roberts, 1980) si nota che per riscaldamento a 150 K l'acqua è completamente desorbita. Se si preadsorbe ossigeno e poi acqua, a 80 K si notano (v. fig. 17a) i picchi dovuti agli ossigeni delle due specie, O(ads) e H2O(ads). Per successivo riscaldamento l'evoluzione è rappresentata nella fig. 17b, che mostra ancora la presenza di H2O a 153 K, 17c, che mette in evidenza la formazione della nuova specie OH(ads), e infine 17d, dove si nota la riduzione ma non la scomparsa della specie formata a 173 K (v. fig. 17c) per reazione superficiale O(ads) + H2O(ads) = 2 OH(ads).
c) Risonanza di spin elettronico (ESR).
Un breve cenno all'impiego della risonanza di spin elettronico, generalmente nota come ‛spettroscopia ESR' (Electron Spin Resonance) o EPR (Electron Paramagnetic Resonance), può essere utile per illustrare qualche esempio di problemi e risultati nel campo della chimica delle superfici (v. Delgass e altri, 1979).
La tecnica FSR si basa sulle transizioni, indotte da un campo magnetico oscillante, tra stati elettronici a spin parallelo e a spin antiparallelo rispetto a un campo magnetico esterno: è quindi necessario che esistano specie aventi elettroni non appaiati (elettroni ‛dispari'). La condizione è necessaria ma non sufficiente: solo in alcuni casi e in alcune condizioni dette specie possono essere ‛viste'. A questa limitazione fa riscontro una grandissima sensibilità della tecnica, che permette di ‛vedere' una concentrazione molto bassa, fino a circa 1013 spin/cm3. La spettroscopia ESR quindi può essere di valido aiuto anche nella chimica delle superfici, in quanto può registrare l'esistenza di specie presenti sulla superficie.
Vi sono sostanzialmente due tipi di problemi in cui si applica la ESR: a) la caratterizzazione di specie appartenenti alla superficie del solido e responsabili della sua interazione con molecole esterne; b) la caratterizzazione di specie adsorbite o formate a seguito di adsorbimento.
Rientra nel primo caso l'individuazione di stati di ossidazione anomali di ioni cataliticamente attivi, stabilizzati dalle particolari condizioni esistenti alla superficie: per esempio, si individuano ioni quali Cr5+, Mo5+, Ni3+, ecc. Rientrano nel secondo caso gli studi sull'adsorbimento di specie molecolari su centri donatori o accettori di elettroni, con formazione rispettivamente di radicali anionici o cationici, o sulla loro interazione con specie adsorbite donatrici o accettrici di elettroni: per esempio, dal CO2 si può formare CO2-, dal nitrobenzene, NB, lo ione radicale NB-, ecc.
La teoria mostra che lo spettro fornisce indicazioni anche sulle interazioni tra specie paramagnetiche, sulla loro concentrazione, sulla simmetria dei siti. L'applicazione della tecnica ESR non si limita all'identificazione ma, se possibile, si estende allo studio delle condizioni che favoriscono la creazione delle specie paramagnetiche, la loro evoluzione nel tempo e il loro comportamento con reagenti.
Un'importante classe di specie superficiali è quella delle specie derivate dall'ossigeno, dato il loro possibile ruolo in reazioni di ossidazione. Sono state messe in evidenza su superfici varie specie, quali O-, O2-, O3-. È anche stato studiato il ruolo che particolari siti elettron-donatori, dovuti per esempio alla presenza di ioni di metalli di transizione, possono avere sia sulla formazione delle specie ossidiche di superficie, sia sulla reattività in presenza di CO. È da notare che la superficie stabilizza alcune forme, non presenti all'interno di un cristallo.
6. Nota conclusiva.
A chiusura dell'articolo può essere utile mettere in evidenza alcune caratteristiche della chimica delle superfici e le prospettive di questa disciplina.
Va sottolineata la continuità tra fisica e chimica delle superfici; in particolare quest'ultima si è basata sui progressi teorici e sperimentali della fisica, e la sua identità emerge soprattutto dai tipi di problemi affrontati. È da tener presente che la soluzione di problemi chimici richiede spesso un compromesso tra rigore e fattibilità; basti pensare al fatto che la chimica delle superfici deve spesso prendere in esame solidi policristallini, non esenti da impurezze, immersi in ambiente fluido che interagisce con la superficie in modo spesso non controllato. Queste condizioni si contrappongono al caso tipico studiato dalla fisica delle superfici, che tende a operare su monocristalli e su superfici definite sia per la morfologia che per la composizione.
Si è anche citata la caratteristica di procedere per problemi e la interdisciplinarità della chimica delle superfici: ciò comporta una continua evoluzione delle tecniche impiegate, di cui qualche esempio è stato dato nell'articolo. È prevedibile che queste tendenze si accentuino nel futuro. All'interno del vasto campo della scienza dei materiali, centrale per il nostro tempo, la chimica delle superfici ha un suo posto non tra i minori.
Bibliografia.
Barr, T.L., Applications of electron spectroscopy to heterogeneous catalysis, in Practical surface analysis by Auger and X-ray photoelectron spectroscopy (a cura di D. Briggs e M. P. Seah), Chichester 1983, pp. 283-358.
Biloen, P., Sachtler, W.M.H., Mechanism of hydrocarbon synthesis over Fischer-Tropsch catalysts, in ‟Advances in catalysis", 1981, XXX, pp. 165-216.
Boccuzzi, F., Garrone, E., Zecchina, A., Bossi, A., Camia, M., Infrared study of ZnO surface properties. II. H2-CO interaction at room temperature, in ‟Journal of catalysis", 1978, LI, pp. 160-168.
Breck, D.W., Zeolite molecular sieves, New York 1974.
Briggs, D., Seah, M.P. (a cura di), Practical surface analysis by Auger and X-ray photoelectron spectroscopy, Chichester 1983.
Che, M., Tench, A.J., Characterization and reactivity of molecular oxygen species on oxide surfaces, in ‟Advances in catalysis", 1983, XXXII, pp. 1-148.
Cimino, A., X-ray photoelectron spectroscopy (XPS) analysis of oxide solid solutions, in ‟Materials chemistry and physics", 1985, XIII, pp. 221-241.
Cimino, A., Carrà, S., Chemisorption and catalysis on metal oxides, in Electrodes of conductive metallic oxides (a cura di S. Trasatti), Amsterdam 1980, pp. 97-140.
Clark, A., The theory of adsorption and catalysis, New York 1970.
Defay, R., Prigogine, I., Belleman, A., Everett, D.H., Surface tension and adsorption, London 1966.
Delgass, W.N., Haller, G.L., Kellerman, R., Lunsford, J.H., Spectroscopy in heterogeneous catalysis, New York 1979.
Dowden, D.A., Crystal and ligand field models of solid catalysts, in ‟Catalysis reviews", 1971, V, pp. 1-32.
Flood, E.A. (a cura di), The solid gas interface, New York 1967.
García-Moliner, F., The band picture in the electronic theories of chemisorption on semiconductors, in ‟Catalysis reviews", 1968, II, pp. 1-66.
Gates, B.C., Katzer, J., Schuit, G.C.A., Chemistry of catalytic processes, New York 1979.
Knözinger, H., Ratnasamy, P., Catalytic aluminas: surface models and characterization of surface sites, in ‟Catalysis review", 1978, XVII, pp. 31-70.
Morrison, S.R., The chemical physics of surfaces, New York 1977.
Rhodin, T.N., Ertl, G. (a cura di), The nature of the surface chemical bond, Amsterdam 1979.
Rhodin, T.N., Gadzuk, J.W., Electron spectroscopy and surface chemical bonding, in The nature of the surface chemical bond (a cura di T.N. Rhodin e G. Ertl), Amsterdam 1979, pp. 113-273.
Roberts, M.W., Photoelectron spectroscopy and surface chemistry, in ‟Advances in catalysis", 1980, XXIX, pp. 55-95.
Sachtler, W.M.H., Surface composition of alloys, in ‟Applications of surface science", 1984, XIX, pp. 167-180.
Sachtler, W.M.H., Plank, P. van der, The role of individual surface atomsin chemisorption and catalysis by nickel-copper alloys, in ‟Surface science", 1969, XVIII, pp. 62-79.
Sachtler, W.M.H., Santen, R.A. van, Surface composition and selectivity of alloy catalysis, in ‟Advances in catalysis", 1977, XXVI, pp. 69-119.
Somorjai, G.A., Chemistry in two dimensions: surfaces, Ithaca, N.Y., 1981.
Swalin, R.A., Thermodynamics of solids, New York 19723.
Tanabe, K., Solid acids and bases, New York 1970.
Willis, R.F., Lucas, A.A., Mahan, G.D., Vibrational properties of adsorbed molecules, in The chemical physics of solid surfaces and heterogeneous catalysis, vol. II, Adsorption at solid surfaces (a cura di D.A.King e D.P. Woodruff), Amsterdam 1983, pp. 59-163.
Wolkenstein, Th., The electronic theory of catalysis on semiconductors, Oxford 1963.
Wynblatt, P., Ku, R.C., Surface energy and solute strain energy effects in surface segregation, in ‟Surface science", 1977, LXV, pp. 511-531.