
sclerosi tuberosa
sclerosi tuberosa
Complesso di condizioni ereditarie caratterizzate da amartomi e tumori benigni dell’encefalo e di altri organi. I geni responsabili della malattia sono sui cromosomi 9 e 16. La prima individuazione della malattia risale a Désiré-Magloire Bourneville che, nel 1880, ne coniò il nome, tanto che la patologia è nota anche con l’eponimo di malattia di Bourneville.
Sintomatologia e diagnosi
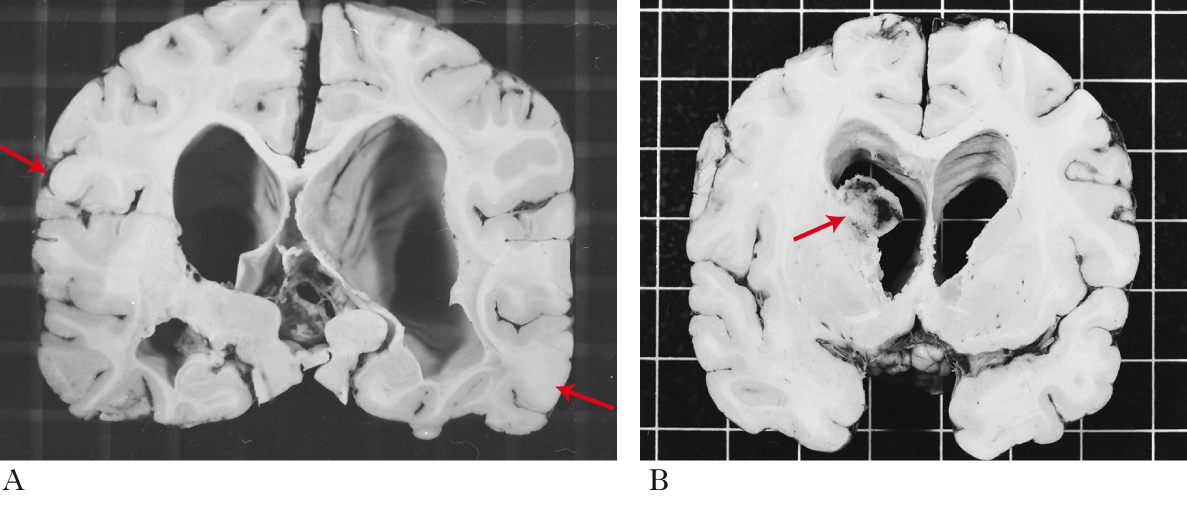
La triade classica dei sintomi comprende epilessia, adenoma sebaceo, ritardo mentale. Si stima una prevalenza di 10÷16 casi per 100.000 neonati e di 7÷12 casi per 100.000 persone; circa la metà dei casi non è diagnosticata. La maggior parte dei pazienti presenta sintomi nella prima decade di vita. I sintomi neurologici sono i più frequenti e in genere gravi: epilessia, spasmi infantili e disturbi psichiatrici quali autismo e ritardo mentale. La diagnosi si basa sugli aspetti clinici, che sono quanto mai variabili da caso a caso. Si distinguono manifestazioni maggiori e manifestazioni minori, la cui combinazione produce la diagnosi di s. t. certa, probabile e possibile. Le manifestazioni maggiori più frequenti sono a carico del sistema nervoso e della pelle. L’astrocitoma gigante subependimale, i tuberi corticali (amartomi corticali e sottocorticali), i noduli subependimali le eterotopie della sostanza bianca sono presenti nella quasi totalità dei pazienti. Si tratta di lesioni tumorali cerebrali con scarsissima propensione alla crescita che producono epilessia o sindrome da ipertensione endocranica. Istologicamente sono costituite da cellule dismorfiche con fenotipo variabile gliale o neuronale, al limite tra la natura tumorale e quella malformativa. Le manifestazioni cutanee tipiche sono l’angiofibroma della faccia (detto anche adenoma sebaceo), le macchie ipomelanotiche e la cosiddetta peau de chagrin (Shagreen patches), pelle zigrinata. L’angiofibroma della faccia produce macchie o papule rossastre sul naso e sulle guance, con distribuzione a farfalla, ed è costituito da vasi sanguigni e tessuto fibroso. Le macchie ipomelanotiche, da assenza di melanina, possono trovarsi in qualunque parte del corpo; in genere sono il solo segno visibile di malattia alla nascita. La peau de chagrin consiste in zone di pelle ruvida e ispessita che si presentano nella regione lombare e alla nuca; il nome deriva dalla somiglianza con la pelle usata anticamente per rilegare i libri e derivata da un asino selvatico, detto onagro (chagrin in francese). Manifestazioni maggiori meno frequenti sono gli amartomi retinici, gli angiomiolipomi renali, i rabdomiomi (amartomi miofibromatosi) cardiaci, gli amartomi epatici, la linfangioleiomiomatosi polmonare. Quest’ultima è una proliferazione disordinata di cellule muscolari lisce nei bronchioli, setti alveolari e spazi linfatici del polmone, che produce ostruzione delle piccole vie aeree, formazione di pneumotorace e di cisti polmonari, fino alla sostituzione del parenchima polmonare con un agglomerato multicistico; l’impegno delle vie linfatiche può portare a versamento di chilo nella pleura. Il rabdomioma del cuore è un rischio di insufficienza cardiaca solo nel feto e nel neonato. La presenza di due manifestazioni maggiori porta alla diagnosi di s. t. certa. Se ne è presente solo una, la diagnosi richiede l’esame genetico, anche se non si dispone di marcatori efficienti e nel 15% degli individui con segni clinici non si trova la mutazione.
Eziologia genetica
L’ereditarietà è autosomica dominante, ad alta penetranza ma con grande variabilità fenotipica. Una storia famigliare di malattia è evidente solo nel 50% dei casi; questo indica un’alta frequenza di mutazioni nuove. Due sono i siti genici alternativamente implicati: uno è sul cromosoma 9 (Tsc1), l’altro sul cromosoma 16 (Tsc2). Il prodotto genico di Tsc1 si chiama amartina ed è una proteina delle vescicole citoplasmatiche, a funzione imprecisata; è fortemente espressa nel tessuto nervoso, nel cuore e nel rene, organi frequentemente implicati nella malattia. Mutazioni producenti una proteina troncata sono state trovate nel 13% dei pazienti di famiglie diverse. Tsc2 codifica una proteina detta tuberina diffusamente espressa in molti tessuti. Tsc2 è associato a forme più gravi della malattia; il riscontro di mutazioni di Tsc2 nei pazienti è più alto che quello di mutazioni di Tsc1. A tutt’oggi (2010) non sono emerse correlazioni tra evento genico e fenotipo clinico. Esistono evidenze che tuberina e amartina agiscono stabilmente in modo sinergico, sia in vitro sia in vivo, costituendo un complesso coinvolto nel controllo della divisione cellulare; di conseguenza, le mutazioni risultano in perdita del controllo e tendenza a formare tumori. Si può quindi ipotizzare che i due geni della malattia agiscano come geni oncosoppressori.
Prognosi
La prognosi dipende dal carico e dalla gravità di segni e sintomi, che possono variare da minime manifestazioni cutanee a gravi disabilità psichiatriche e compromissione neurologica, epilessia, insufficienza renale. Tuttavia, per lo più le manifestazioni sono trattabili con terapie sintomatiche e quindi l’aspettativa di vita dei pazienti è normale. Se l’evento genico è noto, è possibile la sorveglianza nella famiglia e, anche se ardua, la diagnosi prenatale. Cause di morte dei pazienti sono le affezioni renali, i tumori cerebrali, lo stato di male epilettico. Sono in corso sperimentazioni (2010) sull’uomo riguardanti l’efficacia della rapamicina, inibitore di un sistema cellulare favorente la proliferazione, che si è rivelato in grado di indurre la regressione dei tumori cerebrali in un modello murino di sclerosi tuberosa.