
Sangue
Sangue
(XXX, p. 664; App. II, ii, p. 782; III, ii, p. 657; IV, iii, p. 261; V, iv, p. 621)
Nell'Enciclopedia Italiana e nelle Appendici gli argomenti relativi all'ematologia hanno trovato ampia trattazione: gli aspetti di fisiologia e patologia del s. sono svolti in voci specifiche e soprattutto nella voce sangue che nell'App. V è dedicata interamente agli sviluppi delle tecniche trasfusionali. Sempre nell'App. V sono inseriti argomenti di grande rilevanza svolti nelle voci emoglobinopatie (ii, p. 94), emopatie (ii, p. 97) ed emopoiesi (ii, p. 99). In questa Appendice la trattazione della voce sangue è riservata ad argomenti di fisiologia e di patologia nei quali si sono realizzati recentemente grandi progressi: la coagulazione del s., le coagulopatie e le malattie tumorali. Quest'ultimo contributo aggiorna puntualmente la voce emopatie dell'App. V (e in particolare le leucemie e i linfomi) e in conclusione descrive dettagliatamente i trapianti di midollo osseo e inserisce un cenno all'impiego di terapie biologiche, come la terapia genica e l'immunoterapia. *
Coagulazione del sangue
di Giuseppe Maria Gandolfo, Laura Conti
Moderna interpretazione dell'attivazione del sistema della coagulazione
Per coagulazione del s. si intende l'espressione funzionale del complesso sistema di proteine plasmatiche che, in condizioni fisiologiche, permette l'arresto delle emorragie attraverso la formazione del reticolo di fibrina.
Negli ultimi tre lustri del 20° sec. le nostre conoscenze sulla fisiologia del sistema della coagulazione si sono notevolmente ampliate, modificando la concezione 'classica' del processo di attivazione del sistema. Si riteneva infatti in passato che tale processo si verificasse 'a cascata', cioè per la successiva trasformazione di proenzimi in enzimi. In realtà le reazioni di attivazione sono più complicate, richiedendo spesso la formazione di complessi attivanti, comprendenti enzimi, cofattori plasmatici, fosfolipidi e ioni calcio. Si riteneva inoltre che il processo di attivazione potesse seguire due vie, una intrinseca e una estrinseca: nella prima sarebbero intervenute proteine già presenti nel plasma e attivate inizialmente dalle superfici non endoteliali; la seconda avrebbe invece avuto avvio per intervento della tromboplastina tessutale o fattore tessutale, normalmente assente nel plasma.
Tale interpretazione è stata modificata in base a osservazioni di natura sperimentale, biochimica e clinica. È stato così dimostrato che l'attivazione della coagulazione inizia sempre per intervento del fattore tessutale e prosegue con una serie di reazioni non più schematicamente separabili in due sequenze. È in tal modo comprensibile perché le carenze congenite dei primi fattori della cosiddetta via intrinseca (fattori xii, xi) non provochino lo sviluppo di gravi sindromi emorragiche, e perché i difetti di fattori, come l'viii o il vii, che interverrebbero esclusivamente nella via intrinseca o in quella estrinseca, non siano compensati in vivo dalla normale attivazione dell'altra via.
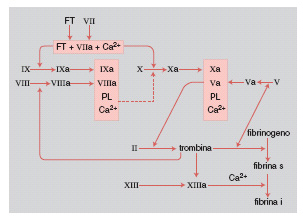
L'attivazione del sistema della coagulazione del s. avviene attraverso sei tappe principali (fig. 1):
1) formazione del complesso fattore tessutale + viia + Ca²⁺: quando il fattore tessutale FT fosfoglicoproteina presente su tutte le membrane cellulari, a eccezione di quella piastrinica, viene esposto all'esterno delle cellule o comunque immesso in circolo, si forma il complesso FT + viia + Ca²⁺, che ha la proprietà di attivare due reazioni: a) fattore ix → fattore ixa; b) fattore x → fattore xa;
2) formazione del complesso ixa + viiia + fosfolipidi (PL) + Ca²⁺: una volta attivato il fattore ix, in presenza del fattore viii (la cui persistenza in circolo dipende dalla contemporanea presenza del fattore von Willebrand, e che viene attivato da tracce di trombina), di fosfolipidi (forniti fisiologicamente dalla membrana piastrinica) e di ioni calcio, si forma il complesso ixa + viiia + PL + Ca²⁺ che, come il complesso FT + viia + Ca²⁺, ha la proprietà di attivare il fattore x;
3) formazione del fattore xa: entrambi i complessi finora esaminati hanno la capacità di operare la trasformazione del fattore x in xa. Questa è pertanto una fase essenziale del processo di attivazione della coagulazione, nella quale confluiscono sia la sequenza principale, a opera del fattore tessutale, sia la sequenza amplificata, ma non meno importante, con intervento del fattore ixa;
4) formazione del complesso xa + va + PL + Ca²⁺: una volta attivato il fattore x, si forma il complesso xa + va (attivato da tracce di trombina) + PL + Ca²⁺, detto anche protrombinasi, in quanto capace di attivare la protrombina;
5) formazione della trombina: la protrombina, glicoproteina formata da un'unica catena polipeptidica, viene trasformata dal fattore xa in trombina attraverso una serie di reazioni, alla fine delle quali si forma questo enzima proteolitico;
6) formazione della fibrina: l'azione proteolitica della trombina si svolge essenzialmente sulla molecola del fibrinogeno, costituita da un dimero, ciascuna metà essendo formata da tre catene polipeptidiche A, Bβ, γ ; la trombina stacca un frammento peptidico da ciascuna catena A (fibrinopeptide A) e da ciascuna catena Bβ (fibrinopeptide B). Si forma così il monomero di fibrina. I diversi monomeri di fibrina quindi, sempre in presenza di trombina, si aggregano formando un reticolo di fibrina solubile, così detta in quanto ancora facilmente dissociabile. Infine, in presenza di trombina e del fattore xiii, avviene una reazione di transamidazione tra i singoli monomeri, con formazione della definitiva fibrina insolubile.
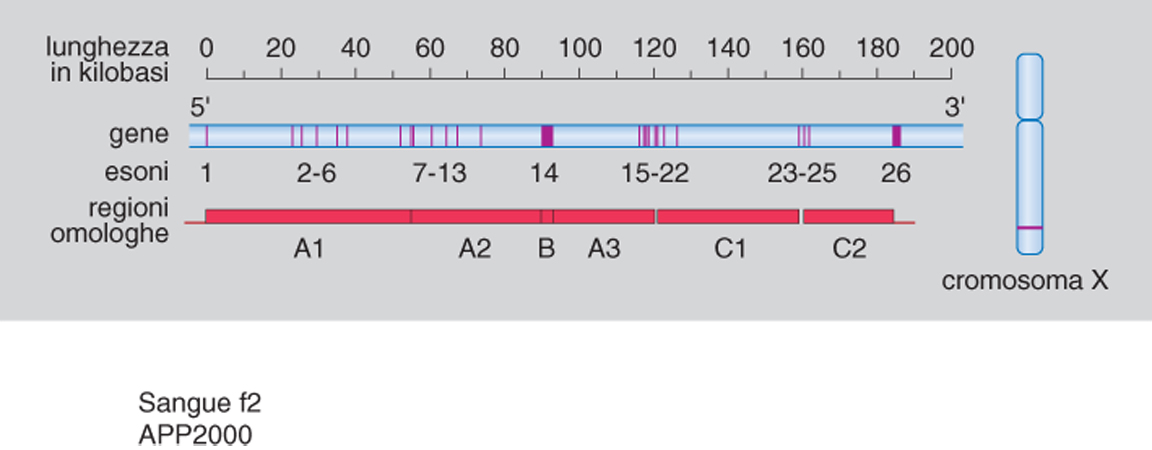
Questo breve cenno sui complessi fenomeni di attivazione del sistema della coagulazione riflette solo in minima parte gli enormi sviluppi che sono stati ottenuti in questi ultimi anni in tale branca dell'ematologia. I progressi hanno riguardato gli aspetti di genetica molecolare degli emoderivati sempre più attivi e sicuri. In questa sede si ricorda soltanto che gli studi di genetica molecolare hanno permesso di chiarire meglio alcuni aspetti etiopatogenetici dell'emofilia A (la coagulopatia congenita più grave) e di formulare sia una corretta diagnosi prenatale sia una precisa diagnosi di portatrice di anomalia genetica correlata all'emofilia A. Il gene del fattore viii, che costituisce lo 0,1% del cromosoma X, è una complessa struttura nucleotidica formata da 26 esoni, 25 introni, e 186.000 paia di basi (fig. 2) Il fattore viii, a sua volta, è formato da 2351 aminoacidi, di cui 19 costituiscono il peptide secretore del precursore del fattore viii. Nei pazienti con emofilia A gli studi di genetica molecolare hanno dimostrato la presenza sia di delezioni, sia di inserzioni, sia di mutazioni puntiformi, rivelate spesso dall'enzima di restrizione TaqI e coinvolgenti il dinucleotide CpQ (citosina-guanina).
L'applicazione di metodiche di genetica molecolare ha poi permesso di identificare in modo certo le portatrici dell'anomalia del cromosoma X e di effettuare diagnosi prenatale mediante prelievi bioptici di villi coriali all'8a÷12a settimana di gestazione. In questi casi si può ottenere un'identificazione indiretta del difetto (mediante lo studio dell'associazione di tale difetto con polimorfismi genetici strettamente adiacenti al gene del fattore viii) o un'identificazione diretta (quando il difetto genetico è ben evidente, come nel caso di una grossa delezione). Nella maggior parte dei casi si applica la prima procedura, che del resto viene utilizzata anche nella diagnostica di altre malattie ereditarie. In pratica il DNA, estratto dai leucociti del s. periferico, viene digerito da enzimi di restrizione che riconoscono specifiche sequenze polimorfe: si formano così frammenti di variabile lunghezza (RFLP, Restriction Fragment Length Polymorphism; v. biologia molecolare, in App. V e in questa Appendice) presenti anche in vicinanza del gene in studio. Questi RFLP vengono trasmessi da una generazione all'altra associati al gene stesso con un comportamento allelico e quindi, nell'ambito di una data famiglia, possono essere impiegati come marcatori per l'identificazione del gene mutato. Nell'emofilia A, in relazione alla grandezza del gene e alla variabilità dei difetti molecolari osservati, sono stati identificati diversi polimorfismi correlati al gene per il fattore viii, permettendo così una diagnosi genetica in oltre il 90% delle famiglie studiate.
La regolazione del processo emocoagulativo da parte del sistema della proteina C anticoagulante
Era noto da molti anni che il processo di attivazione fisiologica della coagulazione viene controllato da una serie di proteine inibitrici le varie fasi di questo processo. In realtà, è nota da tempo la fondamentale azione dell'antitrombina iii, inibitore fisiologico capace di inattivare tutti i fattori attivati della coagulazione che agiscono come enzimi di tipo serina-proteasico (trombina, fattori xa, ixa, xia), e la cui carenza allo stato omozigote è incompatibile con la vita fetale, mentre allo stato eterozigote provoca una spiccata tendenza a trombosi venose profonde già in età giovanile.
Negli ultimi anni l'analisi del sistema degli inibitori fisiologici della coagulazione si è notevolmente ampliata grazie allo sviluppo delle conoscenze sul cosiddetto sistema della proteina C anticoagulante.
La proteina C anticoagulante è un proenzima, vitamina K-dipendente, che viene attivato (APC, Activated Protein C) in un enzima di tipo serina-proteasico dalla trombina legata al suo recettore endoteliale, la trombomodulina. L'APC degrada i fattori va e viiia, insieme alla proteina S (anch'essa vitamina K-dipendente) libera, cioè non legata alla proteina che trasporta anche la frazione C4b del sistema del complemento; la proteina S agisce come cofattore non enzimatico, favorendo il legame dell'APC alle superfici fosfolipidiche, e inibisce direttamente l'attivazione della protrombina interagendo con i fattori va e viiia. Di recente è stato dimostrato che anche il fattore v agisce come cofattore dell'APC, in quanto coopera con la proteina S nel potenziamento della degradazione del fattore viiia e forse del fattore va a opera della proteina C attivata.
L'analisi del sistema della proteina C anticoagulante ha permesso di identificare le basi molecolari di una frequente causa di trombosi eredofamiliare: era stato infatti casualmente osservato che il plasma di alcuni pazienti con trombosi eredofamiliari non presentava, dopo aggiunta di proteina C attivata, un allungamento del tempo di tromboplastina parziale, come quello dei soggetti normali ('resistenza alla proteina C attivata'). Tale anomalia, trasmessa come carattere autosomico dominante, presente nel 2÷15% delle popolazioni occidentali e causa più frequente (fino al 40÷50%) di tutte le trombosi eredofamiliari giovanili, è in realtà dovuta a una mutazione (G→A in posizione 1691) del gene del fattore v, con conseguente sostituzione di un'arginina in posizione 506 con un residuo di glutamina. Questo 'fattore v mutato' (FV gene mutation, chiamato anche fattore v Leiden) è particolarmente resistente all'azione inibitoria svolta dalla proteina C attivata, ed è quindi responsabile di trombosi recidivanti.
bibliografia
J. Gitschier, W.I. Wood, T. M. Goralka et al., Characterization of human factor viii gene, in Nature, 1984, 312, pp. 326-30.
G.M. Gandolfo, L. Conti, La malattia trombotica, Roma 1992.
P.J. Fay, Factor viii structure and function, in Thrombosis Haemostasis, 1993, 70, pp. 63-67.
H.R. Robert, Molecular biology of hemophilia B, in Thrombosis Haemostasis, 1993, 70, pp. 1-9.
C.T. Esmon, W. Ding, K. Yasuhiro et al., The protein C pathway: new insights, in Thrombosis Haemostasis, 1997, 78, pp. 70-74.
Coagulopatie
di Giuseppe Maria Gandolfo, Laura Conti
Per coagulopatie si intendono tutte quelle condizioni patologiche congenite o acquisite nelle quali la carenza di uno o più fattori della coagulazione è responsabile dello sviluppo di sindromi emorragiche. Gli sviluppi delle nostre conoscenze in questi ultimi anni a tal riguardo sono stati enormi e hanno interessato la caratterizzazione di alcune forme congenite (in particolare della malattia di von Willebrand), la diagnostica sia delle forme congenite sia delle forme acquisite, anche grazie a tecniche di biologia molecolare, la preparazione di emoderivati sempre più sicuri e di recente, con la tecnica del DNA ricombinante, le basi per lo sviluppo della terapia genica nei deficit congeniti dei fattori della coagulazione del sangue.
La caratterizzazione delle varianti della malattia di von Willebrand
La malattia di von Willebrand, la più frequente carenza congenita di un fattore della coagulazione, è una malattia emorragica ereditaria, trasmessa come carattere autosomico, dovuta alla difettosa sintesi del fattore von Willebrand, proteina ad alto peso molecolare, che fisiologicamente svolge due funzioni: permette l'adesione delle piastrine alle strutture sottoendoteliali, in particolare al collagene, e costituisce la proteina di trasporto per il fattore viii che, in assenza del fattore von Willebrand, avrebbe un'emivita di poche ore.
Il gene del fattore von Willebrand si trova sul cromosoma 12, contiene 6 esoni e 5 introni; nei soggetti con malattia di von Willebrand sono stati descritti diversi tipi di delezione del gene del fattore von Willebrand. Da tempo è noto che i pazienti con malattia di von Willebrand presentano una multiforme sintomatologia emorragica (fondamentalmente a livello della cute e delle mucose) e una variabilità dei reperti di laboratorio che permettono di porre la corretta diagnosi, come l'allungamento del tempo di emorragia, l'allungamento del tempo di tromboplastina parziale, il deficit di fattore viii, il deficit di fattore von Willebrand, la difettosa adesione delle piastrine, la ridotta o assente aggregazione piastrinica provocata dalla ristocetina.
Attualmente si conosce la caratteristica struttura monomerica del fattore von Willebrand, presente sotto forma di triplette nel plasma e di doppiette nelle piastrine; inoltre nel plasma normale sono presenti dei polimeri di fattore von Willebrand sia ad alto peso molecolare sia a basso peso molecolare. In realtà la malattia di von Willebrand comprende diverse anomalie genetiche, capaci di modificare in vario modo la sintesi di tale fattore, per cui, alla fine, la struttura del fattore von Willebrand nei vari sottogruppi di pazienti può essere diversa, con conseguenti differenze per quanto riguarda la sintomatologia clinica e la semeiotica funzionale. Sono state così identificate diverse varianti, biologicamente differenti, la cui caratterizzazione è indispensabile in quanto il trattamento farmacologico (a base di desmopressina, derivato della vasopressina, capace di aumentare la concentrazione del fattore von Willebrand nel plasma) e sostitutivo (con emoderivati contenenti il fattore von Willebrand) è differente nelle singole varianti.
Emofilia A: basi molecolari; diagnostica molecolare di portatrice; diagnosi prenatale. - Il gene del fattore viii è molto grande, superando i 186 kb, posti in una coppia di 1 megabase sul telomero del braccio lungo del cromosoma X. La regione che codifica il fattore viii è di circa 7300 nucleotidi, contenuti in 26 esoni.
Gli studi di genetica molecolare hanno dimostrato che, in realtà, nei diversi pazienti, con forme gravi, moderate o lievi di emofilia A, si riscontrano vari tipi di mutazioni, come sostituzioni di nucleotidi, mutazioni non-senso, delezioni. Alcune recenti osservazioni fanno ritenere possibile che modificazioni a carico dell'introne 22 siano particolarmente frequenti, almeno nelle popolazioni di origine caucasica. Questa enorme variabilità delle mutazioni nell'emofilia A rende praticamente impossibile una diagnosi molecolare diretta sul DNA, sia per quanto riguarda la condizione di portatrice, sia per quanto riguarda la diagnosi prenatale. Per questi scopi si è pertanto costretti a utilizzare l'analisi dei polimorfismi del DNA correlati al gene per il fattore viii (v. sopra).
Utilizzando particolari enzimi di restrizione, è possibile individuare polimorfismi intragenici o extragenici che permettono la diagnosi di portatrice con un errore del 2÷6%, mentre in circa il 40% di diagnosi prenatali effettuate dal 1993 a oggi è stato possibile identificare con certezza le condizioni genetiche del feto, cioè se era affetto o meno da emofilia A. Nei prossimi anni non sembra impossibile raggiungere il 100% di accuratezza nella diagnosi prenatale.
Trattamento sostitutivo dei difetti congeniti della coagulazione con emoderivati ricombinanti
Il primo prodotto preparato con la tecnica del DNA ricombinante è stato l'insulina umana, nel 1982. Da allora, a scopo clinico, sono state preparate almeno altre 20 proteine, compresi i fattori della coagulazione del s. viii, ix, vii e von Willebrand.
Nel 1988 sono iniziati i primi studi clinici controllati sull'impiego del fattore viii ricombinante, ottenuto da colture cellulari e somministrato in prodotti contenenti l'albumina umana. Questi studi hanno dimostrato che tali emoderivati sono efficaci come il fattore viii purificato da plasma normale, sono sicuri in quanto non trasmettono virus, non provocano l'insorgenza di anticorpi anti-fattore viii in misura maggiore rispetto al fattore viii preparato da plasma normale. Successivamente è stato preparato un fattore viii ricombinante di seconda generazione, caratterizzato dalla delezione di una frazione centrale della catena singola; questo prodotto, denominato r-viii SQ (S per serina, Q per acido glutammico, i due aminoacidi che si uniscono dopo la delezione, Ser 743-Glu 1638), è ugualmente attivo rispetto ai precedenti emoderivati (v. sangue, in App. V), presenta una maggiore attività specifica, è stabile pur essendo privo di albumina umana. Anche per il fattore ix, la cui carenza è causa di emofilia B, è stato possibile preparare di recente un emoderivato ricombinante, prodotto in assenza di proteine umane, con attività indistinguibile da quella del fattore ix plasmatico.
Di recente è stato preparato il fattore viia ricombinante, che è stato già ampiamente utilizzato nei pazienti affetti da emofilia A o emofilia B, nei quali si sia sviluppato un inibitore diretto rispettivamente contro il fattore viii o contro il fattore ix, tale da rendere inefficace la classica terapia sostitutiva con emoderivati specifici. Il fattore viia ricombinante è risultato efficace nel controllo degli episodi emorragici e nella prevenzione delle emorragie postoperatorie di questi pazienti emofiliaci, e contemporaneamente meno trombogenico rispetto agli emoderivati contenenti i fattori attivati del complesso protrombinico (ii, vii, ix, x), utilizzati nel medesimo tipo di pazienti. Infine sono in corso studi preliminari sull'impiego del fattore von Willebrand ricombinante, stabile senza aggiunta di albumina, capace di aumentare, negli animali con deficit di tale fattore, la concentrazione del fattore viii, normalizzando i suoi livelli per molte ore, e controllando le emorragie spontanee dovute alla carenza di fattore von Willebrand.
Terapia genica dell'emofilia. - La possibilità di ottenere una guarigione definitiva attraverso la terapia genica (v. App. V), come già si tenta per altre malattie ereditarie, è oggetto di studio da alcuni anni in modelli animali. Con i recenti sviluppi nella tecnologia del trasferimento dei geni, è stato possibile ottenere l'espressione di fattore viii umano in animali normali e in topi o cani emofiliaci. Tuttavia l'espressione in vivo del fattore viii è risultata molto variabile, da un giorno a oltre 5 mesi. I prossimi sviluppi nel miglioramento dei vettori del trasferimento dei geni e dei metodi che permettono l'espressione dei fattori della coagulazione potranno costituire in futuro la base per un'efficace realizzazione della terapia genica anche nei pazienti con emofilia A o con emofilia B.
bibliografia
H.H. Kazazian, The molecular basis of hemophilia A and the present status of carrier and antenatal diagnosis of the disease, in Thrombosis Haemostasis, 1993, 70, pp. 60-62.
L.M. Aledort, Von Willebrand disease: from the bedside to therapy, in Thrombosis Haemostasis, 1997, 78, pp. 562-65.
E. Berntorp, Second generation, B-domain deleted recombinant factor viii, in Thrombosis Haemostasis, 1997, 78, pp. 256-60.
S. Connelly, M. Kaleko, Gene therapy oh hemophilia A, in Thrombosis Haemostasis, 1997, 78, pp. 31-36.
R.C. Eisensmith, S.L.C. Woo, Viral vector-mediated gene therapy for hemophilia B, in Thrombosis Haemostasis, 1997, 78, pp. 24-30.
H.P. Schwarz, P.L. Turecek et al., Recombinant von Willebrand factor, in Thrombosis Haemostasis, 1997, 78, pp. 571-76.
G.C. White, A. Beebe, B. Nielsen, Recombinant factor ix, in Thrombosis Haemostasis, 1997, 78, pp. 261-65.
Malattie tumorali
di Franco Mandelli, Giovanna Meloni
Tra le malattie tumorali del s., le leucemie e i linfomi rappresentano le forme più frequenti e quelle riguardo alle quali durante gli ultimi anni si sono ottenuti i maggiori progressi dal punto di vista della ricerca di base, con conseguenti miglioramenti nell'approccio diagnostico e nei risultati terapeutici. Qui di seguito vengono presi in esame: le leucemie acute, la leucemia mieloide cronica, la leucemia linfatica cronica, il morbo di Hodgkin, i linfomi non Hodgkin. Viene quindi trattato l'impiego terapeutico del trapianto di midollo osseo allogenico e autologo, con particolare riguardo ai risultati clinici ottenuti nelle neoplasie ematologiche e, nell'ultima parte, si analizzano la terapia immunologica e la terapia genica.
Leucemie acute
Le leucemie acute sono di due tipi: linfoidi e mieloidi. Le prime sono più frequenti nei bambini (circa l'80% dei casi), mentre le seconde prevalgono negli adulti e negli anziani (più del 50% dei casi in pazienti di età superiore ai 60 anni). Per le mieloidi acute l'incidenza è di circa 2 nuovi casi l'anno su 100.000 soggetti; per le linfoidi acute l'incidenza è simile, il che corrisponde a circa 600 nuovi casi l'anno in Italia nei bambini di età inferiore a 15 anni, e a circa 200 casi per i soggetti di età superiore ai 15 anni.
Etiopatogenesi. - L'etiologia delle leucemie acute è tuttora sconosciuta: è stato comunque osservato l'aumento dell'incidenza nei soggetti esposti a radiazioni ionizzanti, o a seguito di incidenti nucleari o per motivi professionali, e nei pazienti sottoposti a terapie con farmaci antineoplastici, soprattutto gli alchilanti. Anche fattori ambientali, in particolare il benzene e derivati, possono aumentare l'incidenza delle leucemie. Dev'essere inoltre tenuto presente che alcune condizioni genetiche, come per es. la sindrome di Down, si accompagnano a una maggiore incidenza di leucemie. Nel processo della leucemogenesi sono state acquisite nuove conoscenze, anche se molti aspetti sono ancora da chiarire. A livello cromosomico sono state identificate numerose alterazioni, sia nelle forme linfoidi sia in quelle mieloidi; è importante sottolineare che alcune di queste alterazioni sono specifiche di varietà come la traslocazione 15:17 nella leucemia mieloide promielocitica, o la traslocazione 8:14 nella linfoide a immunofenotipo B maturo, e che il loro riconoscimento può essere molto utile sotto il profilo diagnostico e terapeutico. Numerose alterazioni cromosomiche interessano i siti dove sono stati identificati alcuni oncogèni che sono senz'altro coinvolti nel processo di leucemogenesi.
Il coinvolgimento di geni specifici per determinate leucemie (per es., nella leucemia promielocitica, una varietà di leucemia mieloide acuta, è coinvolto il gene del recettore dell'acido retinoico) può consentire nuove strategie terapeutiche mirate nei confronti del clone neoplastico, con possibilità di straordinari risultati come quelli che si ottengono con l'impiego dell'acido retinoico (ATRA, All-Trans-Retinoic Acid) nella leucemia promielocitica (v. oltre).
Diagnosi. - Le leucemie acute possono presentarsi con una sintomatologia molto grave, dovuta sia a insufficiente produzione di globuli rossi, piastrine e granulociti da parte del midollo, con la comparsa rispettivamente di anemia, sindrome emorragica e infezioni, sia a localizzazione della malattia leucemica con possibile interessamento di tutti gli organi e apparati (linfonodi, milza, fegato, sistema nervoso centrale, cute ecc.); sia infine alla liberazione di sostanze tossiche da parte delle cellule leucemiche con febbre più o meno elevata e astenia profonda. La sintomatologia può essere molto grave - tale da richiedere l'immediato ricovero del malato per la terapia del caso - o di lievissima entità, o addirittura assente. Sempre più spesso la diagnosi viene effettuata in pazienti del tutto asintomatici, sulla base dei risultati degli esami di laboratorio, soprattutto quello emocromocitometrico, eseguiti in controlli casuali. In corso di leucemia acuta nel s. periferico, oltre alla presenza di anemia, piastrinopenia o granulocitopenia più o meno grave, vi può essere un aumento del numero dei globuli bianchi, fino a valori anche molto elevati (superiori a 100.000/mm³), per la comparsa in circolo di cellule leucemiche.
L'accertamento diagnostico, che nei casi con presenza di elementi leucemici nel s. periferico è molto facile, richiede sempre la valutazione del midollo osseo mediante aspirato midollare, che può essere eseguito a livello del manubrio sternale e/o delle ossa iliache.
Strategia terapeutica nelle leucemie acute. - Diagnosticata e caratterizzata la varietà di leucemia acuta, dev'essere stabilita la strategia terapeutica, che, nella fase di attacco, deve portare alla scomparsa della maggioranza delle cellule leucemiche presenti in tutto l'organismo, compreso il midollo osseo. Se il risultato è ottimale si ottiene la cosiddetta remissione completa, che consiste nella scomparsa di tutti i sintomi clinici soggettivi e obiettivi legati alla malattia e nella normalizzazione dei valori dei globuli rossi, dei globuli bianchi, delle piastrine e del midollo osseo.
Si parla di remissione completa e non di guarigione, in quanto il risultato ottenuto corrisponde alla spiccata riduzione (oltre il 99%) della massa leucemica, ma non alla sua scomparsa; infatti, mentre all'esame microscopico non sono più riconoscibili le cellule leucemiche, con l'impiego di tecniche più sensibili (fenotipo immunologico, citogenetica, biologia molecolare) è sempre possibile dimostrarne la persistenza come malattia minima residua.
Durante la terapia di attacco, per la mancata attività selettiva dei farmaci nei confronti delle cellule leucemiche, si ha un aggravamento dell'anemia, della piastrinopenia e soprattutto della granulocitopenia dovuto all'ulteriore riduzione a livello midollare dei precursori ematopoietici normali (aplasia midollare terapeutica). È in questa fase che, soprattutto nelle leucemie mieloidi acute, si devono attuare tutti i presidi diretti a prevenire e a combattere le complicanze del trattamento antileucemico, rappresentate essenzialmente da emorragie e da infezioni gravi, talvolta mortali.
La probabilità di conseguire la remissione completa dipende dalla varietà di leucemia e dall'età del paziente: si ottiene, infatti, in oltre il 95% dei bambini e in più dell'80% degli adulti con leucemia linfatica acuta, mentre nelle leucemie mieloidi acute si ha in circa l'80% dei bambini e nel 65÷70% degli adulti. Nei pazienti di età superiore ai 60÷65 anni la probabilità di remissione è inferiore - circa il 50% - in entrambe le varietà di leucemia acuta.
Molto recentemente nella leucemia promielocitica la prognosi è radicalmente cambiata rispetto a qualche anno fa, quando questa era ritenuta la forma più grave di tutte le leucemie acute. In tale variante di leucemia, indipendentemente dall'età, da quando viene impiegato in associazione alla chemioterapia l'acido retinoico (ATRA) si ottiene con la terapia di attacco una percentuale di remissione completa molto elevata (circa il 90% dei casi).
Dopo la remissione, tutti i pazienti affetti da leucemia acuta devono essere curati con la cosiddetta terapia postremissionale; tale trattamento - come quello d'attacco - utilizza più farmaci (polichemioterapia) dotati di meccanismi d'azione diversi. La polichemioterapia è diversa nelle forme linfoidi rispetto a quelle mieloidi: più protratta (per almeno due anni) nelle leucemie linfoidi, più breve (circa 6 mesi) e più intensa nelle leucemie mieloidi. Per quanto riguarda le forme linfoidi va tenuto presente che, per la frequente comparsa di localizzazioni a livello del sistema nervoso (meningosi leucemica), dev'essere effettuato un trattamento specifico per la loro prevenzione, trattamento che prevede la somministrazione di farmaci per via intratecale, associata a radioterapia craniale o ad alte dosi di farmaci per via endovenosa che oltrepassano la barriera emato-encefalica.
Circa il 60% dei bambini con leucemia linfoide acuta e il 25÷30% degli adulti ha la probabilità di diventare un lungo sopravvivente senza recidiva, e quindi di guarire. Nelle leucemie mieloidi acute la probabilità di guarigione è minore (circa il 35÷40% dei bambini e il 20÷25% degli adulti). Fa sempre eccezione la leucemia promielocitica, in cui, con una terapia postremissionale intensa e di breve durata, la probabilità di guarigione è superiore al 50% dei casi, sia nei bambini sia negli adulti.
Nonostante la terapia postremissionale, nella maggior parte dei pazienti - con percentuali diverse nelle varie forme di leucemia acuta e nelle diverse età - si verifica la ricomparsa della malattia, cioè la recidiva leucemica. La prima recidiva dev'essere curata in modo intensivo allo scopo di ottenere una nuova remissione che, se persistente, permetterà ancora al paziente di guarire. In questa fase di malattia più avanzata trova indicazione elettiva, se le condizioni generali del paziente lo permettono, un trattamento intensivo ad alte dosi, seguito da infusione di cellule staminali (procedura trapiantologica). Tale trattamento verrà discusso in seguito.
Leucemia mieloide cronica
La leucemia mieloide cronica, rara nei bambini e più frequente nell'adulto e nell'anziano con un'età mediana di circa 50 anni, ha un'incidenza di un nuovo caso ogni 100.000 soggetti all'anno, per cui in Italia vi sono circa 600 nuovi casi all'anno. È dovuta alla trasformazione neoplastica della cellula staminale ed è caratterizzata dalla presenza a livello cromosomico di una specifica alterazione presente in oltre il 95% dei casi, il cromosoma Philadelphia (Ph1). Si tratta della prima specifica alterazione cromosomica dimostrata in una malattia neoplastica, consistente in una traslocazione reciproca tra il cromosoma 9 e il cromosoma 22, con la formazione a livello molecolare di un gene ibrido, dovuto alla fusione dell'oncogène ABL (v. oncogeni, in App. V e in questa Appendice), localizzato nel cromosoma 9, con la regione BCR, localizzata nelle braccia lunghe del cromosoma 22.
La diagnosi della malattia in circa il 30% dei casi è casuale; in pazienti del tutto asintomatici viene dimostrato un aumento del numero dei globuli bianchi, talvolta anche superiore ai 100.000/mm³. L'alterazione numerica dei globuli bianchi si accompagna a un aumento percentuale e assoluto degli elementi granulocitari con comparsa in circolo di elementi nelle diverse fasi di maturazione.
Dal punto di vista dell'obiettività clinica si può rilevare un aumento di volume del fegato e della milza, talvolta molto spiccato. Fondamentale per la diagnosi è lo studio del midollo osseo, non tanto per la spiccata prevalenza degli elementi della serie granulocitaria a livello citomorfologico, quanto per la dimostrazione del cromosoma Ph1, anomalia citogenetica caratteristica.
Il decorso della malattia è in genere cronico e, con le terapie attuali, la sopravvivenza media è di 4÷5 anni. Quasi sempre la morte avviene per la trasformazione della malattia in una forma acuta (crisi blastica), caratterizzata da una sintomatologia molto più grave e da referti di laboratorio e midollari sovrapponibili a quelli delle leucemie acute. La diagnosi di crisi blastica è basata sul riscontro a livello del s. periferico e del midollo di un'elevata percentuale di elementi leucemici molto più immaturi.
Terapia. - Il trattamento di elezione della leucemia mieloide cronica è basato, quando possibile, sul trapianto di midollo allogenico. Nei pazienti anziani, in quelli senza donatore compatibile e in quelli a prognosi più favorevole trova attualmente indicazione l'interferone (IFN), un farmaco - dotato di attività inibente la mielopoiesi - di cui non è stata ancora dimostrata un'attività citotossica selettiva nei confronti dell'emopoiesi Ph1 positiva. L'attività antiproliferativa dell'interferone si esplica favorendo, con meccanismi ancora non chiari, la ripresa dell'emopoiesi normale. L'interferone, rispetto ad altri farmaci antileucemici di tipo chemioterapico, si è dimostrato in grado di aumentare in modo significativo la sopravvivenza media dei pazienti con leucemia mieloide cronica. L'efficacia clinica è particolarmente evidente non solo nei pazienti che, sottoposti a terapia con interferone, ottengono un controllo clinico ed ematologico della malattia, ma soprattutto in quelli in cui si ha una risposta citogenetica. È stato infatti dimostrato che, a differenza di altri trattamenti basati sulla chemioterapia, l'interferone consente di ottenere una risposta citogenetica con una diminuzione percentuale delle cellule Ph1, diminuzione che può arrivare fino alla negativizzazione completa. Tale risultato, confermato in diversi studi pluricentrici in tutto il mondo, consente di affermare che la terapia non trapiantologica deve sempre impiegare l'interferone come farmaco fondamentale.
Dopo crisi blastica, la prognosi nella leucemia mieloide cronica è infausta a breve scadenza, fatta eccezione per i casi in cui, con un trattamento polichemioterapico simile a quello impiegato nelle leucemie acute, si riesca a ottenere il ripristino della fase cronica che consente di eseguire successivamente il trapianto midollare.
Leucemia linfatica cronica
La leucemia linfatica cronica è la più frequente emopatia neoplastica dell'adulto nel mondo occidentale, con un'incidenza di 2÷3 nuovi casi l'anno per 100.000 individui, per cui in Italia vi sono circa 1500 nuovi casi all'anno. È rara al di sotto dei 40 anni; la massima incidenza si ha nei soggetti ultrasessantenni.
La diagnosi viene effettuata in più del 50% dei casi in corso di esami di laboratorio routinari. Il rilievo di un aumento del numero dei globuli bianchi dovuto a una linfocitosi assoluta in persone del tutto asintomatiche porta ad approfondire l'accertamento diagnostico. Nella leucemia linfatica cronica, come nelle leucemie acute, tutti gli organi e apparati possono essere coinvolti dal processo leucemico, anche se in genere questo aspetto si verifica nelle fasi più avanzate. Gli esami di laboratorio dimostrano costantemente una linfocitosi assoluta, che può superare 100.000 linfociti/mm³. Nelle forme più gravi può essere presente un'insufficienza midollare con anemia e/o piastrinopenia; in questi casi a livello del midollo osseo è evidente una sostituzione del normale tessuto emopoietico da parte dei linfociti leucemici che devono essere studiati dal punto di vista immunologico per dimostrarne il carattere monoclonale.
Il decorso è spesso lento, con una durata media di sopravvivenza che supera comunque i cinque anni. Vi sono pazienti che per l'assenza di sintomi non richiedono alcun trattamento anche per anni, e muoiono per cause diverse dalla malattia. Altri pazienti presentano un andamento più aggressivo e devono essere trattati precocemente, talvolta già all'esordio. La maggioranza dei pazienti, soprattutto se di età superiore ai 60÷65 anni, viene curata con terapie molto blande, effettuabili a domicilio, praticamente prive di tossicità, che consentono di controllare il processo patologico per molti anni con una buona qualità di vita.
Più difficile è la scelta terapeutica nei pazienti giovani, con forme di leucemia linfatica cronica ad andamento più aggressivo, per cercare di modificare il decorso irreversibilmente fatale della malattia. In questi pazienti vengono utilizzati farmaci nuovi, quali gli analoghi delle purine, che hanno una forte tossicità, con possibile comparsa di complicanze emorragiche e soprattutto infettive, anche mortali, analoghe a quelle che si osservano durante la terapia aggressiva che viene impiegata nelle leucemie acute. Nella leucemia linfatica cronica vengono attualmente utilizzate con risultati estremamente interessanti, pur se del tutto preliminari, le terapie intensive includenti il trapianto autologo o il trapianto allogenico.
Morbo di Hodgkin
Il morbo di Hodgkin è una malattia con un'incidenza di 0,5 casi su 100.000 individui all'anno, per cui in Italia vi sono circa 500 nuovi pazienti all'anno. La malattia colpisce tutte le età della vita, con due picchi di maggiore incidenza, uno tra 20 e 30 anni e uno oltre i 50 anni. Per la etiopatogenesi è stato recentemente ipotizzato il coinvolgimento del virus di Epstein e Barr, agente etiologico della mononucleosi infettiva, non solo per l'aumentato rischio per i pazienti con mononucleosi infettiva di sviluppare il morbo di Hodgkin e per il riscontro nei pazienti con morbo di Hodgkin di elevati titoli anticorpali per il virus, ma soprattutto perché è stata dimostrata la presenza di acidi nucleici o di proteine virali nei tessuti coinvolti dalla malattia. Resta comunque da chiarire se il virus di Epstein e Barr rappresenti un semplice ospite, o possa essere invece veramente implicato nella patogenesi della malattia.
Questa può esordire con sintomi sistemici (febbre, sudorazione, dimagrimento) o, più frequentemente, con linfoadenopatie che possono essere localizzate sia alle stazioni superficiali sia a quelle profonde. In alcuni casi la diagnosi risulta molto difficile, in quanto può essere presente come unico sintomo un prurito incoercibile, talora generalizzato. Le localizzazioni della malattia possono coinvolgere tutti gli organi e apparati, mentre più raramente vi è un quadro di insufficienza midollare dovuto a un coinvolgimento del midollo osseo. Il quadro di laboratorio non è caratteristico e non consente mai la diagnosi, che è sempre basata sull'esame istopatologico del tessuto neoplastico mediante biopsia mirata di un linfonodo superficiale o con un intervento chirurgico più complesso.
In questa malattia è fondamentale la stadiazione, cioè la valutazione della sua estensione, che dev'essere fatta non solo sulla base di dati obiettivi, ma soprattutto sulla base di accertamenti radiografici (radiologia tradizionale, tomografia assiale computerizzata, risonanza magnetica nucleare). Una corretta stadiazione è importante non solo ai fini prognostici, ma anche per impostare la terapia. Nelle forme localizzate, senza sintomi sistemici, il trattamento è basato sull'impiego della radioterapia, mentre nelle forme più diffuse e in quelle con segni sistemici svolge un ruolo fondamentale la polichemioterapia. Generalmente comunque il trattamento dev'essere associato: radio e chemioterapico.
Nella stragrande maggioranza dei pazienti dopo la terapia si ottiene la remissione completa, con la scomparsa di tutti i sintomi e della localizzazione della malattia, non solo a livello obiettivo, ma anche a livello radiologico. Dopo la remissione e la sospensione del trattamento il paziente viene sottoposto a controlli periodici per una valutazione a lungo termine dell'efficacia terapeutica e per poter effettuare una diagnosi precoce in caso di un'eventuale recidiva. Con una corretta strategia terapeutica più del 60% dei pazienti guarisce con il primo trattamento; comunque anche dopo recidiva è possibile ottenere nuovamente la remissione, che si può accompagnare a lunga sopravvivenza in più del 30÷40% dei pazienti se successivamente vengono sottoposti a terapia eradicante.
Linfomi non Hodgkin
I linfomi non Hodgkin si distinguono essenzialmente in due gruppi, quelli a basso grado di malignità, molto simili come caratteristiche cliniche alla leucemia linfatica cronica, e quelli ad alto grado di malignità che, viceversa, possono avere caratteristiche biologico-cliniche, almeno in alcune varietà, simili a quelle delle leucemie linfoidi acute. L'incidenza dei linfomi non Hodgkin è di circa 6 casi su 100.000 individui l'anno, per cui in Italia se ne diagnosticano circa 3000 nuovi casi l'anno. Le forme ad alto grado di malignità possono colpire tutte le età della vita, mentre le forme a basso grado prevalgono nei soggetti di età superiore ai 50 anni e sono assenti nei bambini. Nella trasformazione neoplastica delle cellule bersaglio, cioè dei linfociti, svolgono un ruolo importante le anomalie genetiche.
La sintomatologia di esordio è estremamente eterogenea, con la possibile comparsa di un aumento dei linfonodi superficiali e profondi come unico sintomo; talvolta l'ingrandimento interessa una sola stazione linfonodale, ma più spesso (a differenza del morbo di Hodgkin) sono interessati linfonodi in più sedi. La milza e il fegato possono essere spesso sede di malattia già all'esordio. In un terzo circa dei pazienti alla diagnosi può essere presente un interessamento extralinfonodale talvolta come unica sede di malattia (cute, apparato gastroenterico, sistema nervoso centrale, polmoni, midollo). Anche nei linfomi non Hodgkin, soprattutto ad alto grado di malignità, possono essere presenti segni sistemici, con astenia marcata, febbre, sudorazione, dimagrimento; il prurito invece è del tutto eccezionale. Nel s. periferico, se la malattia invade il midollo osseo si può avere anemia, granulocitopenia e piastrinopenia con la presenza nei linfomi leucemici di cellule linfomatose nel s. periferico.
Per la diagnosi è fondamentale l'accertamento istologico con prelievo bioptico del tessuto colpito, che consente non solo di distinguere le forme a basso o ad alto grado di malignità, ma anche di identificare i vari sottotipi istologici con importanti riflessi prognostico-terapeutici.
La prognosi e la terapia debbono essere poste in relazione non solo con la varietà istologica e con l'età, ma anche con lo stadio, l'interessamento extralinfonodale, con parametri di laboratorio fra cui la latticodeidrogenasi e soprattutto con l'invasione midollare e con la leucemizzazione. Nei linfomi a basso grado di malignità ad andamento cronico, spesso non è necessaria alcuna terapia e sono sufficienti trattamenti blandi che possono garantire una normale qualità di vita anche per molti anni. Nei linfomi a basso grado di malignità ad andamento aggressivo e in quelli ad alto grado di malignità deve sempre essere rapidamente instaurata una polichemioterapia, che in genere viene effettuata per pochi mesi e che si associa nella maggior parte dei casi a una remissione completa. Nei linfomi a basso grado di malignità, tale remissione si protrae senza recidiva in un'elevata percentuale di casi, con una probabilità di guarigione in circa il 50% dei casi. Nei linfomi ad alto grado di malignità, si riesce quasi sempre a ottenere una remissione clinica parziale o completa, che in genere però non è protratta nel tempo, in quanto è documentabile nella maggior parte dei pazienti la persistenza di malattia a livello genetico molecolare (malattia minima residua). Per questo motivo sono in corso di valutazione nei pazienti più giovani trattamenti potenzialmente eradicanti, associati a trapianto di midollo osseo. Dopo recidiva dev'essere attuata una terapia intensiva seguita da autotrapianto in tutti i casi eleggibili.
Un aspetto di estremo interesse è rappresentato dall'approccio diagnostico-terapeutico dei linfomi, quasi sempre ad alto grado di malignità ed estremamente aggressivi, che compaiono in corso di infezione HIV, talvolta come primo segno di AIDS. Le attuali terapie dell'infezione da HIV, che consentono un buon controllo non solo dell'infezione virale, ma anche delle complicanze a essa legate con un netto aumento della sopravvivenza, hanno cambiato la prognosi anche in questa particolare varietà di linfomi. Queste patologie, sino a pochi anni fa invariabilmente e rapidamente fatali, attualmente, se trattate in modo intensivo includente anche le terapie trapiantologiche, possono non solo regredire completamente, con l'ottenimento di una remissione completa, ma anche essere eradicate con guarigione della patologia linfomatosa.
Procedure trapiantologiche
di Franco Mandelli, Giovanna Meloni
L'intensità di dose, cioè la dose di farmaco (antiblastico) somministrata per unità di tempo, rappresenta attualmente un aspetto fondamentale nella strategia terapeutica delle leucemie e dei linfomi. Da oltre vent'anni nelle neoplasie ematologiche viene effettuato il trapianto di midollo allogenico, che prevede prima del trapianto l'impiego di radiochemioterapie sovramassimali (regimi di condizionamento), allo scopo di ottenere una completa eradicazione della malattia con una maggiore probabilità di guarigione. I regimi di condizionamento, per la tossicità ematologica proibitiva che altrimenti determinerebbero, sono utilizzabili solo se seguiti dall'infusione di cellule staminali, non solo del donatore normale (allotrapianto) ma anche dello stesso paziente (autotrapianto). Tale infusione può consentire di ricostituire una emopoiesi normale.
La reinfusione delle cellule staminali permette quindi di utilizzare dosi sovraletali di radio e/o chemioterapia, superando i limiti dovuti alla tossicità midollare, allo scopo di aumentare l'efficacia del trattamento: l'unico fattore limitante è rappresentato dalla tossicità a carico di altri organi o tessuti. Più recentemente, la possibilità di ottenere un numero elevato di cellule staminali dal s. periferico del paziente o del donatore normale, utilizzando metodiche basate soprattutto sulla somministrazione dei fattori di crescita emopoietici, ha consentito l'impiego della terapia sovramassimale con infusione di cellule staminali da s. periferico (CSP) nella maggior parte delle neoplasie ematologiche, con un numero progressivamente crescente di pazienti trattati in tutto il mondo.
Si distinguono quindi due tipi di trapianto di cellule staminali: quello con reinfusione di cellule autologhe ottenute dallo stesso paziente e quello con reinfusione di cellule allogeniche di un donatore normale. Come donatori devono essere utilizzati preferibilmente i fratelli, o, in assenza di fratelli compatibili, donatori di midollo volontari che devono essere perfettamente compatibili con il ricevente; più recentemente - ma si tratta di una terapia ancora sperimentale - vengono impiegate anche le cellule staminali contenute nel s. del cordone ombelicale, sempre a condizione che siano compatibili con il ricevente.
L'allotrapianto è un vero trapianto e presenta aspetti negativi e positivi. I primi sono legati, oltre che alla reazione del trapianto verso l'ospite (GVHD, Graft Versus Host Disease, v. anche oltre; con la necessità di eseguire una terapia immunosoppressiva) - reazione che può insorgere immediatamente dopo l'infusione cellulare (forme acute) o più tardivamente (forme croniche) -, alla notevole incidenza di complicanze infettive soprattutto virali e all'impiego di regimi di condizionamento più immunosoppressivi, con notevole rischio di complicanze infettive tardive. Gli aspetti positivi sono rappresentati da un effetto da parte delle cellule immunocompetenti del donatore nei confronti della malattia leucemica (reazione contro la leucemia) e ovviamente dall'assenza del rischio legato alla contaminazione delle cellule staminali da reinfondere da parte di cellule neoplastiche, contaminazione che può essere presente se si reinfondono cellule staminali autologhe. Accanto al classico prelievo di cellule midollari, che dev'essere eseguito in anestesia generale o epidurale prelevando il midollo osseo dalle creste iliache posteriori, il s. periferico è sempre più largamente utilizzato come fonte di cellule staminali, grazie all'impiego dei separatori cellulari a flusso continuo o discontinuo.
Con l'impiego in associazione della chemioterapia e dei fattori di crescita emopoietici, il numero di aferesi (raccolta selettiva di cellule) necessario per raccogliere un numero di cellule staminali in grado di determinare un buon attecchimento è molto basso (può essere addirittura sufficiente una sola aferesi).
Per quanto riguarda il trapianto da donatore normale, ovviamente viene utilizzato come fattore di crescita il solo GCSF (Granulocyte Colony Stimulating Factor) senza chemioterapia.
Il s. periferico è stato inizialmente utilizzato in combinazione con il midollo osseo per garantire un attecchimento non solo valido, ma anche stabile nel tempo. Le prime esperienze hanno dimostrato la maggiore efficacia dell'associazione del midollo e del s. periferico nei confronti del solo midollo con un più rapido attecchimento per quanto riguarda sia i granulociti neutrofili sia le piastrine. Successivamente è stato ampiamente dimostrato, nelle leucemie acute e croniche e soprattutto nei linfomi, che il s. periferico, da solo, determina, rispetto al midollo, una più rapida ripresa ematologica che persiste nel tempo, come quella che si ottiene con l'infusione di cellule staminali midollari.
Un problema ancora irrisolto, ma sicuramente di estrema importanza, è quello della purificazione delle cellule staminali quando vengono utilizzate le cellule autologhe, cioè quelle dello stesso paziente.
Come è ben noto, le leucemie acute e croniche hanno sempre una diffusione a livello midollare, e anche i linfomi (soprattutto quelli non Hodgkin) possono avere un interessamento midollare. Al momento del prelievo di midollo, la stragrande maggioranza dei pazienti con leucemia ha quindi una più o meno evidente persistenza di malattia, con conseguente rischio di reinfusione di cellule leucemiche, che può avere un impatto nel condizionare l'eventuale recidiva. Questo si può evitare con metodiche di purificazione in vivo o in vitro.
Per quanto riguarda la purificazione in vivo, dopo la remissione e prima di effettuare il prelievo di cellule staminali vengono effettuate ulteriori chemioterapie, dirette a ottenere una sempre più valida remissione completa e una malattia minima residua sempre più piccola.
Per diminuire la contaminazione da parte delle cellule leucemiche o linfomatose sono state utilizzate numerose metodiche di purificazione in vitro. Il requisito ideale per una valida metodica di purificazione in vitro è la scarsa tossicità sulle cellule staminali normali associata a elevata attività citotossica nei confronti delle cellule leucemiche o linfomatose. Sono state utilizzate diverse metodiche immunologiche con l'impiego di anticorpi monoclonali, farmacologiche, o immunologiche e farmacologiche contemporaneamente.
Nelle leucemie mieloidi acute (esclusa la varietà promielocitica la cui prognosi, come abbiamo già detto, è in continuo progressivo miglioramento dopo l'introduzione dell'acido retinoico) la chemioradioterapia sovramassimale, seguita dall'infusione di cellule staminali, sia allogeniche sia autologhe, è stata utilizzata ampiamente in prima remissione completa, sia nei bambini sia negli adulti. I risultati dimostrano chiaramente che la terapia intensiva seguita dall'infusione di cellule staminali allogeniche determina un miglioramento significativo della durata della prima remissione completa; anche il trapianto autologo, sebbene con risultati clinici inferiori a quelli dell'allotrapianto, sembra offrire vantaggi rispetto alla chemioterapia a dosi non sovramassimali. Dopo recidiva, in tutte le varietà di leucemia mieloide acuta, compresa la varietà promielocitica, il trapianto allogenico e quello autologo hanno nei pazienti eleggibili (per età e per condizioni generali) un'indicazione assoluta, con risultati sicuramente migliori di quelli ottenuti con la chemioterapia tradizionale.
Nelle leucemie linfoidi acute dei bambini la terapia sovramassimale associata all'infusione di cellule staminali autologhe può trovare indicazione solo dopo recidiva, soprattutto se extramidollare isolata, con risultati però non particolarmente incoraggianti. Ha invece indicazione assoluta il trapianto allogenico, che peraltro durante la prima remissione ha indicazione soltanto nelle forme ad altissimo rischio di recidiva (elevata massa leucemica all'esordio, anomalie cromosomiche peculiari come alcune traslocazioni).
Nelle leucemie linfoidi acute dell'adulto i risultati, poco favorevoli, ottenuti con la chemioterapia tradizionale hanno stimolato numerosi centri a utilizzare la chemioterapia ad alte dosi con infusione di cellule staminali autologhe e allogeniche in prima remissione completa. L'impiego dell'autotrapianto non ha comunque migliorato in modo significativo i risultati ottenuti con la chemioterapia tradizionale nelle prime remissioni, mentre l'allotrapianto, che ha indicazione assoluta dopo recidiva, si associa a un aumento della sopravvivenza anche se effettuato in prima remissione.
Nella leucemia mieloide cronica il trapianto allogenico ha indicazione assoluta sia dopo trasformazione acuta sia, soprattutto, in fase cronica: si tratta dell'unica terapia che può offrire al paziente la possibilità di guarigione, che si può ottenere in circa il 60% dei casi se il trapianto viene effettuato in fase cronica.
Nella leucemia linfatica cronica l'impiego del trapianto autologo è in fase del tutto preliminare. Sono state comunque già dimostrate la fattibilità e la scarsa tossicità di tale procedura soprattutto se si utilizzano, dopo la terapia ablativa, cellule staminali periferiche. I risultati clinici sono molto incoraggianti, anche se preliminari. Il trapianto allogenico nei casi eleggibili con un donatore compatibile fra i fratelli è l'unica terapia che può determinare la guarigione con una probabilità di circa il 50%, ed è sicuramente la terapia di scelta nei pazienti refrattari alla terapia convenzionale.
Nei linfomi non Hodgkin ad alto grado di malignità la terapia sovramassimale (con infusione di cellule staminali autologhe) è stata ampiamente utilizzata sia nella terapia di induzione della remissione, sia come consolidamento della remissione stessa. Indicazione assoluta ha l'impiego della terapia sovramassimale nei pazienti dopo ricaduta, specie se presentano una malattia sensibile a un nuovo trattamento; ancora da valutare è l'impiego dei trattamenti intensivi nei pazienti che dopo la terapia d'attacco hanno ottenuto soltanto una risposta parziale. Nei linfomi ad alto grado di malignità, è comunque necessario tener presente che i risultati ottenuti nei pazienti de novo con la chemioterapia tradizionale sono molto favorevoli, almeno nei pazienti a rischio non elevato, per cui la terapia sovramassimale dovrà probabilmente essere utilizzata solo in particolari categorie di pazienti a rischio elevato, o nell'ambito di strategie terapeutiche complesse. Il trapianto allogenico ha indicazione soltanto in particolari categorie di pazienti (linfoma linfoblastico) e in quelli resistenti o recidivati dopo trapianto autologo.
Nei linfomi a basso grado di malignità la strategia terapeutica basata sull'impiego delle alte dosi è giustificata, come nella leucemia linfoide cronica, solo nei casi che presentano scarse probabilità di lunga sopravvivenza con la chemioterapia tradizionale. Pochi sono i pazienti in cui è indicato il trapianto allogenico.
Nel morbo di Hodgkin, dopo recidiva rispondente, soprattutto se precoce, la chemioterapia ad alte dosi seguita da infusione di cellule staminali si è dimostrata molto efficace. Sulla base dei risultati favorevoli ottenuti in questo particolare gruppo di pazienti, che con la terapia tradizionale hanno scarsissime possibilità di guarigione, le alte dosi sono state utilizzate precocemente anche come consolidamento della prima remissione nei pazienti ad alto rischio di recidiva. Va comunque tenuto presente che, in questa forma morbosa, i risultati ottenuti nella maggioranza dei casi con la terapia convenzionale e la difficoltà di identificare all'esordio i pazienti a prognosi chiaramente sfavorevole non consentono ancora di stabilire l'esatto valore della terapia ad alte dosi. Anche nel morbo di Hodgkin, data l'elevata tossicità che si accompagna a tale procedura, il trapianto allogenico trova indicazione solo in casi eccezionali.
Fattori di crescita emopoietici
I fattori di crescita emopoietici hanno rivoluzionato la terapia di supporto che tanta rilevanza ha nella strategia terapeutica delle neoplasie ematologiche e non. Un'indicazione consolidata consiste nel loro uso per la mobilizzazione delle cellule staminali nel s. periferico con la possibilità di impiegare, dopo terapia ablativa per l'autotrapianto, in luogo del midollo, le cellule staminali da s. periferico che consentono di ottenere, rispetto alle cellule di origine midollare, un più rapido attecchimento con livelli sufficientemente elevati di neutrofili e di piastrine raggiunti in un periodo più breve. Ciò riduce le complicanze post-trapianto, i giorni di ricovero e quindi i costi della procedura.
Un'altra indicazione è rappresentata dall'impiego dei fattori di crescita emopoietici dopo terapia ad alte dosi, allo scopo di ridurre la durata della neutropenia. I risultati dei numerosi studi disponibili consentono di affermare che l'associazione del fattore di crescita alla chemioterapia determina non solo una minore durata e gravità della granulocitopenia, ma anche una minore incidenza degli episodi febbrili e delle complicanze infettive. Nei pazienti trattati con fattori di crescita è stata dimostrata infatti una riduzione della durata della terapia antibiotica e dei giorni di ricovero ospedaliero.
Terapia biologica
La terapia genica (v. App. V) è una strategia terapeutica che nei prossimi anni potrebbe dare risultati favorevoli in molte forme neoplastiche. L'inserimento di una sequenza genomica nel DNA delle cellule neoplastiche potrebbe, modificando l'espressione di antigeni tumorali specifici, permettere il loro riconoscimento da parte del sistema immunitario. Inoltre l'inserimento nelle cellule neoplastiche di geni soppressori della loro replicazione potrebbe essere utilizzato per arrestarne la crescita.
L'immunoterapia (v. immunopatologia e immunoterapia, in questa Appendice) è una modalità terapeutica basata sulla stimolazione del sistema immunitario del paziente con l'obiettivo di provocare una risposta specifica contro le cellule neoplastiche. I principali effettori del sistema immune sono i linfociti T e le cellule Natural Killer (NK), e la loro attività citotossica è stata confermata da numerosi studi in vitro, su modelli animali e più recentemente da risultati di estremo interesse nell'uomo, soprattutto riguardanti il trapianto allogenico.
Il trapianto di midollo allogenico (v. sopra), anche utilizzando come donatore un fratello completamente compatibile, si accompagna quasi costantemente a un'aggressione da parte del midollo trapiantato nei confronti dell'ospite GVHD (Graft Versus Host Disease). La GVHD comporta una sintomatologia di diversa entità, talvolta pressoché assente, ma altre volte così grave da portare a morte il paziente. La GVHD è stata abolita completamente eliminando i linfociti T nel midollo da reinfondere (T-deplezione), ma ciò ha provocato un aumento spiccato di recidive leucemiche. L'eliminazione dei linfociti T comporta infatti anche l'annullamento del beneficio terapeutico legato alla cosiddetta Graft Versus Leukemia (GVL) dovuta all'attività citotossica dei linfociti T del donatore sulle cellule residue nel paziente, che possono persistere anche dopo terapia di condizionamento.
Esiste la possibilità di potenziare gli effettori del sistema immunitario incrementando la loro attività citotossica nei confronti delle cellule neoplastiche, anche nei pazienti non sottoposti a trapianto allogenico, grazie all'impiego di una citochina come l'interleuchina 2 (IL-2), la cui capacità di proliferazione e potenziamento delle cellule del sistema immunitario e di induzione di attività citotossica è stata valutata in numerosi studi preclinici e clinici. Certamente uno dei campi dove più promettenti sembrano gli impieghi futuri dell'IL-2 è quello ematologico. L'IL-2 è stata sperimentata in diverse patologie neoplastiche ematologiche in diverse fasi di malattia, in remissione e non, e dopo trapianto di midollo autologo. A oggi i risultati più incoraggianti si sono avuti nelle leucemie mieloidi acute in recidiva.
bibliografia
A.J. Barrett, M.M. Horowitz, R.P. Gale et al., Marrow transplantation for acute lymphoblastic leukemia: factors affecting relapse and survival, in Blood, 1989, 74, pp. 862-71.
R.P. Gale, Acute myelogenous leukemia: progress and controversies, in Leukemias, 1990, 4, pp. 529-31.
N.C. Gorin, P. Aegerter, B. Auvert et al., Autologous bone marrow transplantation for acute myelogenous leukemia in I remission, in Blood, 1990, 75, pp. 1606-14.
W.J. Urba, D.L. Longo, Hodgkin's disease, in New England journal of medicine, 1992, 326, pp. 678-87.
American Society of Clinical Oncology, Recommendations for the use of hematopoietic colony stimulating factors: evidence-based, clinical practice guidelines, in Journal of clinical oncology, 1994, 12, pp. 2471-2508.
R.Y. Wells, W.G. Woods, J.D. Buckley et al., Treatment of newly diagnosed children and adolescents with acute myeloid leukemia. A Children Cancer Group study, in Journal of clinical oncology, 1994, 12, pp. 2367-77.
B. Coiffier, Y. Baston, F. Berger et al., Prognostic factors in follicular lymphomas, in Seminars of oncology, 1995, 20, pp. 89-95.
B. Lowenberg, Post-remission treatment of acute myelogenous leukemia, in New England journal of medicine, 1995, 332, pp. 260-62.
E. Montserrat, C. Rozman, Chronic lymphocytic leukemia: present status, in Annals of oncology, 1995, 6, pp. 219-35.
T. Philip, C. Guglielmi, A. Hagenbeek et al., Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non Hodgkin's lymphoma, in New England journal of medicine, 1995, 333, pp. 1540-45.
C.H. Pui, Childhood leukemias, in New England journal of medicine, 1995, 332, pp. 1618-30.
R.A. Zittoun, F. Mandelli, R. Willemze et al., Autologous or allogeneic bone marrow transplantation compared with intensive chemotherapy in acute myelogenous leukemia, in New England journal of medicine, 1995, 332, pp. 217-23.
M. Michallet, E. Archimbaud, G. Bandini et al., HLA-identical sibling bone marrow transplantation in younger patients with chronic lymphocytic leukemia, in Annals of internal medicine, 1996, 124, pp. 311-15.
K. Welte, J. Gabrilove', M.H. Brouchud et al., Filgrastim: the first 10 years, in Blood, 1996, 88, pp. 1907-29.
Acute promyelocytic leukemia. A curable disease, 2nd International symposium, ed. F. Mandelli, Roma 1997.
F.R. Appelbaum, M.M. Horowitz et al., Allogeneic hemopoietic stem cell transplantation for acute leukemia, in Seminars of oncology, 1997, 24, p. 114.
R. Bhratia, C.M. Versaille, J.S. Miller et al., Review autologous transplantation therapy for chronic myelogenous leukemia, in Blood, 1997, 89, pp. 2623-34.
CML Trialists collaborative group, Interferon alpha versus chemotherapy for chronic myeloid leukemia: a metanalysis of seven randomized trials, in Journal of the National Cancer Institute, 1997.
G.F. Laport, R.A. Larson, Treatment of adult acute lymphoblastic leukemia, in Seminars of oncology, 1997, 24, pp. 70-82.
G. Meloni, F.R. Mauro, A. Proia et al., Terapia sovramassimale nella leucemia linfatica cronica, Seminario di Ematologia, Milano 1997, vol. 5°, pp. 79-86.
R. Szydlo, J.M. Goldman, J.P. Klein et al., Results of allogeneic bone marrow transplants using donors other than HLA identical siblings, in Journal of clinical oncology, 1997, 15, pp. 1767-77.
D.Y. Weisdorf, A.L. Billet, P. Hannan et al., Autologous versus unrelated donor allogeneic marrow transplantation for acute lymphoblastic leukemia, in Blood, 1997, 8, pp. 2962-68.