
Sangue
Sangue
Emoglobina, di Austen F. Riggs
Genetica del sangue, di Guido Modiano
Organi emopoietici, di Angelo Baserga e Giovanni Diego Grusovin
Anemie emolitiche, di Corrado Baglioni
Leucemie, di Edoardo Storti e Salvatore Carlo Rizzo
Emoglobina
SOMMARIO: 1. Introduzione. □ 2. Struttura. □ 3. Attività fisiologica dell'emoglobina: a) trasporto dell'ossigeno; legame con l'O2; b) controllo del trasporto di O2 e CO2: effetto Bohr; c) controllo del trasporto di O2 e CO2: fosfati organici. □ 4. Genetica ed evoluzione. □ Bibliografia.
1. Introduzione.
Le emoglobine sono proteine contenenti l'eme, composto tetrapirrolico con ferro (II), cioè ferroso, che possono legare reversibilmente l'ossigeno. Questa definizione distingue le emoglobine dagli enzimi respiratori che contengono l'eme ma non si combinano reversibilmente con l'ossigeno, e include i pigmenti, presenti negli animali invertebrati e spesso indicati come eritrocruorine, e le emoglobine delle cellule dei muscoli, le mioglobine. La funzione primaria delle emoglobine del sangue è il trasporto di ossigeno molecolare. La funzione delle mioglobine sembra consistere nel facilitare la diffusione dell'ossigeno dalla superficie della cellula ai siti di utilizzazione, i mitocondri.
È la parte proteica dell'emoglobina, la globina, che conferisce il carattere di reversibilità alla combinazione dell'eme con l'ossigeno; l'eme di per sé non ha questa capacità e il ferro che esso contiene si ossida irreversibilmente: elettroni sono trasferiti dal metallo all'ossigeno. Quindi la proteina cambia i livelli energetici elettronici dell'atomo di ferro in modo tale che questo trasferimento di carica, nell'ossiemoglobina, diviene facilmente reversibile, cosicché la formazione di ossiemoglobina deve essere considerata più un'ossigenazione che un'ossidazione.
Differenze nella parte proteica sono responsabili dei vari adattamenti fisiologici dell'emoglobina nei diversi animali: spesso l'affinità della proteina per l'ossigeno è correlata con l'efficienza metabolica dell'animale o con la pressione di ossigeno nell'ambiente.
2. Struttura.
Le emoglobine del sangue dei Vertebrati hanno come caratteristica un peso molecolare intorno a 65.000 e sono formate da quattro catene polipeptidiche eguali a due a due, indicate rispettivamente con α e β, cosicché il tetramero può essere indicato come α2β2 (v. fig. 1). La catena α nell'uomo contiene 141 residui di amminoacidi; la catena β ne ha 146. Il feto, nella specie umana, contiene una speciale emoglobina fetale, α2γ2, con catene γ invece di quelle β. Ogni catena polipeptidica avvolge parzialmente un eme, cosicché una molecola tetramerica, contenente quattro catene, può legare 4 molecole di ossigeno. Le emoglobine degli Invertebrati sono di norma o relativamente piccole (peso molecolare 65.000 o meno) e contenute all'interno di cellule, oppure libere, disciolte nel sangue, in cui formano enormi aggregati con pesi molecolari che variano da 400.000 nella pulce d'acqua, Daphnia, a 3×106 in un verme anellide, Arenicola. Questi grossi aggregati possono contenere fino a 200 subunità. La maggior parte di queste emoglobine contiene subunità formate da catene polipeptidiche di peso molecolare 15.000-18.000, ciascuna con un gruppo eme. Tuttavia parecchie emoglobine di Invertebrati, per esempio quelle del verme Ascaris, hanno peso molecolare 35.000-40.000 per subunità. Presumibilmente, o la catena polipeptidica è così grande o vi sono due catene polipeptidiche, una sola delle quali contiene l'eme. Le emoglobine polimeriche possono, in alcuni casi, dissociarsi nelle subunità da cui sono costituite; per esempio le emoglobine dei Mammiferi vanno incontro alla seguente reazione:

Ad alta forza ionica K1 aumenta, mentre K2 diminuisce; quest'ultima è significativa solo a pH inferiore a 5. Anche le grosse emoglobine polimeriche degli Invertebrati vanno incontro a dissociazione in funzione del pH.
Gli eleganti studi di M. F. Perutz e dei suoi collaboratori hanno determinato la struttura sia della desossi- che della ossiemoglobina fino alle dimensioni atomiche. Questa grande impresa è stata realizzata per mezzo dell'analisi di Fourier degli spettri di diffrazione dei raggi X da parte di cristalli di emoglobina. Tali studi hanno mostrato che la conformazione delle singole catene polipeptidiche è molto simile: ciascuna contiene 7-8 segmenti elicoidali (designati A,B,C,D,E,F,G e H, partendo dal segmento A, contenente il gruppo amminico terminale della molecola; v. fig. 2). L'eme si trova tra i segmenti elicoidali E ed F in una sacca idrofobica: l'atomo di ferro è legato covalentemente a un residuo di istidina del segmento F. I gruppi carichi e ionizzabili si trovano, per la maggior parte, all'esterno della molecola e sono accessibili all'ambiente circostante. Nel suo insieme la disposizione di ciascuna catena polipeptidica nelle emoglobine umane e di cavallo è molto simile a quella della mioglobina dei muscoli, la cui struttura è stata determinata da J. C. Kendrew nel 1960. Inoltre anche le emoglobine del verme Glycera e dell'insetto Chironomus presentano nell'insieme una conformazione delle catene molto simile a quella dell'emoglobina umana, malgrado la maggior parte degli amminoacidi siano differenti. È notevole il fatto che si può persino rimpiazzare il ferro con il cobalto e avere ancora una molecola che si combina reversibilmente con l'ossigeno.
Il problema centrale è di determinare le correlazioni tra struttura e funzione, cioè comprendere in che modo la struttura dell'emoglobina sia causa dell'attività fisiologica. Perché la curva di equilibrio dell'emoglobina con l'ossigeno è sigmoide (v. fig. 4)? Poiché la forma a S indica che i monomeri agiscono in modo cooperativo tra di loro, dobbiamo comprendere come l'ossigenazione di una subunità produca un cambiamento dell'affinità per l'ossigeno nelle subunità vicine. Qui i particolari meccanici sono ben lungi dall'essere compresi, benché siano state proposte parecchie teorie plausibili. Un possibile approccio è di considerare il ‛meccanismo scatenante', cioè l'evento, nelle vicinanze dell'atomo di ferro, che porta al cambiamento strutturale. Nell'emoglobina, in assenza di leganti, l'atomo di ferro ha numero di ossidazione +2 ed è paramagnetico e pentacoordinato; quindi la cavità occupata dall'O2 nell'ossiemoglobina è vuota nella desossiemoglobina. La combinazione con l'O2 dà luogo a un composto diamagnetico esacoordinato. L'assenza di proprietà magnetiche nell'ossiemoglobina era stata già messa in evidenza nel 1845 da Faraday, il quale aveva notato che il sangue era l'unica sostanza contenente ferro che non fosse magnetica. L'attacco di un legante, come l'O2 o il CO, sul ferro induce uno spostamento della porfirina rispetto ai segmenti elicoidali E ed F che si trovano ai lati di esso. Un plausibile meccanismo proposto per spiegare questo spostamento è il seguente: il legame con l'istidina della catena F è relativamente fissato a circa 2,0 Å di lunghezza, ma l'atomo di ferro si trova circa 0,8 Å sopra il piano formato dalla porfirina nella desossiemoglobina, mentre giace su questo piano nella ossiemoglobina. Questo spostamento è del tutto sufficiente a generare uno spostamento e una distorsione nelle parti più flessibili della globina. Probabilmente gli spostamenti differiscono, nelle catene α e nelle β, per alcuni dettagli: infatti l'ambiente in cui è localizzato l'eme non è perfettamente identico nei due casi. Nelle catene α sembra esservi abbastanza spazio per accogliere una molecola di ossigeno senza cambiamenti sostanziali nel tratto a elica E, mentre nella catena β vi è un residuo di valina che sporge nella sacca idrofobica quando la proteina è desossigenata; prima di reagire con il legante, evidentemente, la distanza tra la porfirina e l'elica E deve aumentare per consentirne l'ingresso: questo distanziarsi è dovuto a vibrazioni termiche. Si sa che questi spostamenti hanno notevoli ripercussioni in qualche punto di ciascuna subunità, benché i dettagli meccanici di ciò siano molto incerti. Ne risulta, in definitiva, un movimento delle subunità l'una rispetto all'altra; questa può essere una via di ‛comunicazione' di una subunità con l'altra nel produrre il fenomeno di cooperatività nei riguardi del legame con l'ossigeno.
Il rapporto tra questi cambiamenti e l'ordine secondo cui le subunità vengono ossigenate non è ancora noto; sono state in proposito avanzate due opposte teorie. La prima di queste, presentata da J. Monod, J. P. Changeux e J. Wyman, ipotizza che vi siano solo due stati o conformazioni (lo stato R per la forma ossigenata, lo stato T per la desossigenata). Questa teoria prevede che tutte le subunità in un tetramero siano in uno dei due stati: una condizione di ‛tutto o niente'. Invece il modello proposto da Koshland, Nemethy e Filmer prevede che solo la subunità che lega l'ossigeno cambi di conformazione: viene suggerita l'idea dell'‛adattamento indotto', cioè sarebbe l'unione con il legante a indurre il cambio di conformazione in una determinata subunità. Queste idee sono schematizzate nella fig. 3: la prima e l'ultima colonna rappresentano le forme stabili previste nel modello di Monod, mentre la diagonale illustra il modello di Koshland. Dal punto di vista formale lo schema diagonale di Koshland è identico al modello di ossigenazione sequenziale di G. S. Adair (v. sotto, cap. 3). Se tutti i possibili equilibri indicati dalla fig. 3 avessero eguale importanza, la descrizione del legame con l'O2 sarebbe sicuramente troppo complessa dal punto di vista dell'analisi sperimentale; sono state fatte quindi delle ipotesi semplificative per rendere più accessibile il problema. È evidente che l'analisi delle forme pienamente ossigenate o desossigenate, per quanto possa essere completa e minuziosa, di per sé non è sufficiente a chiarire i passaggi avvenuti o gli stati intermedi tra di esse, allo stesso modo che la termodinamica di per sé non dice nulla circa la cinetica di un sistema. Tuttavia una serie di dati più recenti suggerisce che né il modello di Koshland né quello di Monod sono del tutto soddisfacenti e che probabilmente è necessario elaborare un modello composito. Una delle ragioni fondamentali di ciò è che, in alcune condizioni particolari, è possibile ottenere uno stato ‛desossi' anche per l'emoglobina in presenza di leganti e ciò dimostra che gli equilibri rappresentati nell'ultima riga della fig. 3 devono essere importanti. Questa osservazione è in accordo col modello di Monod; il fatto che la dissociazione di protoni dall'emoglobina sia direttamente proporzionale alla quantità di legante presente (effetto Bohr) è d'accordo con il modello di Koshland: il modello di Monod richiederebbe infatti una relazione non lineare.
3. Attività fisiologica dell'emoglobina.
a) Trasporto dell'ossigeno; legame con l'O2.
Già nel 1852 il famoso chimico tedesco J. von Liebig ipotizzò che i globuli rossi del sangue contenessero un composto del ferro che si combinava reversibilmente o con l'ossigeno o col diossido di carbonio. In realtà egli suppose che il composto formato col diossido di carbonio venisse distrutto dall'ossigeno. Tuttavia solo nel 1862 E. F. Hoppe-Seyler dimostrò che era l'emoglobina a possedere la proprietà di legare l'ossigeno, com'era stato suggerito da Liebig. Lo studio del legame dell'emoglobina con l'ossigeno ebbe alterne vicende finché G. Hüfner nel 1901 suppose che la reazione potesse essere semplicemente indicata come Hb+O2⇄HbO2; questo equilibrio può essere descritto con una singola costante:

e dà luogo a una curva iperbolica di saturazione con l'ossigeno
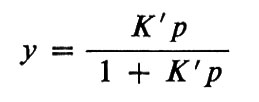
in cui y è la percentuale di saturazione, p è la pressione dell'ossigeno e K′ è la costante di equilibrio; qui la pressione dell'ossigeno è usata invece della concentrazione, a cui è proporzionale. Hüfner determinò K da un solo punto (p, y) su questa curva. Christian Bohr, padre del fisico Niels Bohr, poco soddisfatto da questa scarsa ricerca di punti sperimentali, iniziò una serie di esperimenti per studiare l'effettiva relazione tra y e p e poté dimostrare che la curva di saturazione non era iperbolica come previsto da Hüfner, ma sigmoide (v. fig. 4). Fu A. V. Hill a notare che la forma a 5 poteva essere facilmente spiegata ammettendo che non una, ma diverse molecole d'ossigeno potessero combinarsi con l'emoglobina: Hb+nO2⇄Hb(O2)n; in questo caso la curva di saturazione può essere espressa con

in cui n e K sono costanti. Questa equazione combacia molto bene con i dati sperimentali tra il 10% e il 90% di saturazione (y compreso tra 0,1 e 0,9) e dà una spiegazione adeguata per la maggior parte dei problemi di fisiologia. Hill inizialmente pensò che l'emoglobina dovesse esistere in diversi stati di aggregazione per spiegare il valore di n>1. Nell'equazione è implicito il concetto di cooperatività: il combinarsi di una molecola di ossigeno rende più facile il legarsi delle successive. Il valore sperimentale di n, non intero (2,7-2,8), fu preso come indicazione della presenza di un insieme di molecole di emoglobina legate ognuna con un diverso numero di molecole di ossigeno (1, 2, 3 o 4). Sir J. Barcroft affermò che la curva a S era anomala e cercò a lungo di dimostrare che l'emoglobina nel suo ‛stato nativo' aveva una curva di equilibrio con l'ossigeno a forma d'iperbole. Ma infine prevalse un certo tipo di ragionamento a ritroso: l'emoglobina purificata fu trattata in modo tale da rendere iperbolico il suo equilibrio con l'ossigeno: questa emoglobina così prodotta avrebbe dovuto essere nello stato nativo o ‛primitivo'. Accurate misure di pressione osmotica fatte da Adair nel 1924-1925 dimostrarono che una molecola di emoglobina di cavallo conteneva non uno, ma quattro emi e quindi egli propose che l'ossigenazione dell'emoglobina potesse essere descritta da quattro equilibri successivi:
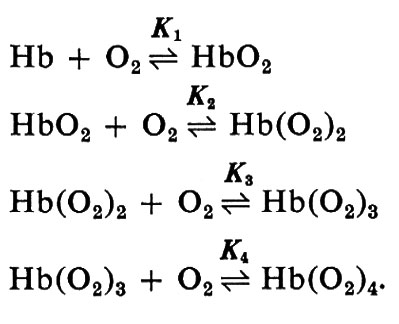
La curva di saturazione che ne risulta è

Benché i dati sperimentali possano essere ottimamente descritti con un'opportuna scelta delle 4 costanti, è da tener presente che praticamente ogni curva semplice può essere descritta con 4 costanti; la possibilità di adattare la curva teorica ai punti sperimentali non dice alcunché circa i meccanismi. Ciò nondimeno lo schema di Adair fornisce un'utile traccia formale.
Non solo l'ossigeno, ma anche il monossido di carbonio si combina rapidamente con l'emoglobina; tuttavia, a differenza dell'ossigeno il CO forma un complesso fotodissociabile. L'affinità dell'emoglobina umana per il CO è circa 200 volte maggiore di quella per l'O2. Ciò si può indicare con l'equilibrio:
HbO2+CO &mis6;Q HbCO+O2
la cui costante di equilibrio è:
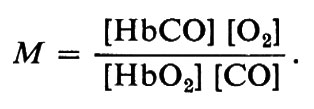
Il valore di M varia tra 50-60 e 400 nelle emoglobine dei Vertebrati, mentre alcune emoglobine di Invertebrati hanno M〈1; quest'ultimo fatto significa che il pigmento ha un'affinità per l'ossigeno maggiore che per il CO. Questo equilibrio e la fotosensibilità di HbCO spiegano perché i soggetti avvelenati da monossido di carbonio devono essere sottoposti alla respirazione di ossigeno puro ed esposti alla piena luce del sole.
Da un punto di vista fisiologico il requisito essenziale del legame dell'ossigeno con l'emoglobina è che questa possa non solo legare l'ossigeno nei polmoni o nelle branchie, ma anche cederlo rapidamente, a pressioni di ossigeno adeguate per il metabolismo cellulare. Quindi dobbiamo considerare, nella curva di saturazione, la regione di ‛carico' e quella di ‛scarico' (v. fig. 4). Tipicamente l'emoglobina nei globuli rossi ossigenati dopo il passaggio attraverso i capillari polmonari è per circa il 95-98% saturata con l'ossigeno (sangue arterioso), mentre il sangue venoso nelle varie regioni varia tra il 25 e il 75% di saturazione. Il valore medio di ossigenazione del sangue che ritorna al cuore dai tessuti è 65-75%: quindi il sangue normalmente cede ai tessuti circa un terzo dell'ossigeno legato. La tensione media di ossigeno alla quale l'emoglobina cede ossigeno ai tessuti spesso sembra adattata alle esigenze metaboliche dell'animale. Così gli animali con metabolismo veloce (per es. il topo) presentano un sangue che cede ossigeno ai loro tessuti a tensioni maggiori di quelle proprie dei grandi mammiferi (per es. l'elefante), che hanno metabolismo lento e nei quali l'ossigeno è ceduto a tensioni inferiori. Questo meccanismo di controllo è funzione dell'affinità dell'emoglobina per l'ossigeno: un'alta affinità significa che l'emoglobina cede l'O2 solo a basse tensioni di O2. Il modo di realizzarsi di questo adattamento è esposto nel capitolo seguente.
b) Controllo del trasporto di O2 e CO2: effetto Bohr.
Christian Bohr e i suoi colleghi, K. A. Hasselbalch e A. Krogh, scoprirono nel 1904 che il CO2 diminuiva l'affinità per l'ossigeno del sangue e dell'emoglobina in soluzione. Questa scoperta era già stata anticipata da diversi scienziati nel secolo precedente: in effetti nel testo Chimica fisiologica scritto nel 1850, C. G. Lehmann notava che il colore del sangue delle rane cambiava da rosso-arancione a porpora quando gli animali erano esposti al diossido di carbonio. Benché in principio si pensasse che l'effetto Bohr fosse dovuto specificamente al CO2, si comprese presto che esso dipendeva da due cause diverse: prima, la trasformazione del CO2 nell'acido corrispondente (H2CO3), e poi la specifica combinazione del CO2 con l'emoglobina. Quindi, l'abbassamento del pH, prodotto dall'H2CO3 o da qualsiasi altro acido, fa diminuire l'affinità dell'emoglobina per l'ossigeno; questo effetto del pH è chiaramente adattativo, in quanto facilita lo scambio sia dell'O2 sia del CO2 nei polmoni e nei tessuti. Consideriamo dapprima i tessuti; in essi il CO2 prodotto va incontro alle seguenti reazioni:
CO2+H2O &mis6;Q H2CO3,
H2CO3 &mis6;Q HCO−3 +H+.
La prima di queste reazioni è catalizzata dall'enzima anidrasi carbonica. Si è anche visto che l'ossiemoglobina è un acido più forte di quanto non sia la desossiemoglobina nell'ambito fisiologico di pH, cosicché la cessione di ossigeno è accompagnata dal legarsi di protoni alla proteina:
H+y HbO2+xH+ &mis6;Q (H+y+x) Hb+O2.
Quindi il CO2, aumentando l'attività degli H+, facilita la dissociazione dell'ossigeno nei capillari dei tessuti. Nei polmoni accade esattamente il processo inverso: l'ossigenazione facilita l'allontanamento del CO2. Questo fenomeno nel suo insieme è noto come ‛effetto Bohr alcalino'. Un fenomeno opposto avviene a pH inferiore a 6,5, in quanto ora la desossiemoglobina diviene un acido più forte dell'ossiemoglobina, ed è detto ‛effetto Bohr acido'. J. Wyman nel 1948 trovò che i dati conosciuti circa le emoglobine di Mammifero potevano essere agevolmente razionalizzati ammettendo l'esistenza di due gruppi acidi per eme, legati all'ossigenazione, uno dei quali diviene più forte (effetto Bohr alcalino), l'altro più debole (effetto Bohr acido) durante l'ossigenazione. Il modo in cui l'equilibrio con l'ossigeno dipende dal pH può essere descritto dall'equazione:
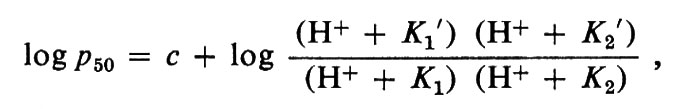
in cui p50 indica la pressione di ossigeno necessaria per il 50% di ossigenazione, K1 e K1′ sono rispettivamente le costanti di dissociazione per il gruppo acido nella ossi- e nella desossiemoglobina, gruppo responsabile dell'effetto Bohr alcalino, mentre K2 e K2′ sono le costanti associate con l'effetto Bohr acido. La costante c ingloba tutti gli altri fattori che influenzano l'affinità per l'ossigeno. Questa equazione descrive il fisiologico effetto Bohr alcalino in termini di un singolo gruppo ionizzabile per eme, ossia 4 gruppi per tetramero. Tuttavia, studi recenti sulla struttura dell'emoglobina mostrano che almeno 6 gruppi ionizzabili per tetramero sono implicati nell'effetto Bohr alcalino: di questi, due sarebbero in ciascuna delle catene α e uno in ciascuna catena β. Quindi, i valori K1 e K1′ riflettono il comportamento risultante di questi gruppi. Inoltre, in presenza di un fosfato organico, il 2,3-difosfoglicerato (DPG), vengono implicati almeno altri due gruppi acidi.
c) Controllo del trasporto di O2 e CO2: fosfati organici.
Per una più profonda comprensione della funzione dell'emoglobina all'interno dei globuli rossi occorre che noi consideriamo questi ultimi come un tessuto in attività metabolica e non solo come sacchetti contenenti emoglobina. Normalmente i globuli rossi di Mammifero, alla temperatura corporea, consumano glucosio a una velocità di 0,3-0,4 mg per ora e per 100 ml di cellule; negli eritrociti nucleati degli Uccelli questa velocità è di circa 10 volte maggiore. Ciascun eritrocita umano contiene circa 280×106 molecole di emoglobina, la quale rappresenta più del 95% di tutte le proteine in esso presenti. Un individuo normale fabbrica eritrociti alla straordinaria velocità di circa 3×106 cellule per secondo, che vengono distrutte alla stessa velocità. Tra sintesi e demolizione, la vita media di un eritrocita è di circa 120 giorni: durante questo periodo le cellule perdono gradualmente alcune delle loro attività enzimatiche e in quelle più vecchie diminuisce l'efficienza nel trasporto di ossigeno. L'insieme di queste attività enzimatiche è essenziale per il mantenimento di un ambiente ottimale per il funzionamento dell'emoglobina e per mantenerne il ferro allo stato ferroso. In condizioni patologiche, nelle quali alcuni di questi enzimi sono assenti o insufficienti, l'emoglobina ferrosa è trasformata gradualmente in ferrica (metemoglobina), e non trasporta più l'O2. La metemoglobina è utile solo nel caso di avvelenamento da cianuro: è noto che una pronta somministrazione, per iniezione, di nitrito di sodio, che ossida l'emoglobina a metemoglobina, può salvare la vita dell'avvelenato; infatti la metemoglobina si lega rapidamente con il cianuro, impedendo che questo reagisca con l'enzima respiratorio, la citocromossidasi.
Le proprietà dell'emoglobina di trasportare ossigeno dipendono dagli ioni intracellulari; per esempio la concentrazione di K+ è generalmente maggiore dentro che fuori l'eritrocita; sia gli ioni K+ sia quelli C- influenzano l'affinità dell'emoglobina per l'ossigeno. Di gran lunga maggiore è l'importanza dei fosfati organici; la concentrazione intracellulare di queste sostanze è regolata metabolicamente: vi è un delicato equilibrio tra il metabolismo degli eritrociti e il trasporto di ossigeno. Ad esempio, la mancanza ereditaria di alcuni enzimi eritrocitari, come la piruvatochinasi e l'esochinasi, è associata con variazioni di affinità del sangue per l'ossigeno: queste variazioni sono in gran parte dovute a variazioni di concentrazione del 2,3-difosfoglicerato (DPG). Come il livello del DPG fa variare il livello di ossigenazione del sangue, così è vero anche il contrario: variazioni dell'ossigenazione fanno variare la concentrazione di DPG. Spesso una diminuzione della saturazione di ossigeno provoca una stimolazione della glicolisi e un accumulo di DPG, e ciò provoca un'ulteriore diminuzione della saturazione di ossigeno. Quindi, sia la permanenza a grandi altitudini, sia una disfunzione cardiaca diminuiscono i livelli di ossigenazione e fanno aumentare il livello del DPG. Recenti studi permettono di avanzare l'ipotesi che l'ormone tiroideo agisca direttamente sull'enzima difosfogliceratomutasi, stimolando la sintesi del DPG.
La rimozione dei fosfati organici con tecniche appropriate, come la dialisi, modifica profondamente la proprietà delle soluzioni di emoglobina. È interessante ricordare che almeno fin dal 1909 è noto che la dialisi altera le proprietà dell'emoglobina; ciò nondimeno questo fenomeno non è stato sistematicamente studiato per più di 50 anni. Ciò è strano, in quanto i biochimici fin dal 1910 avevano scoperto che gli enzimi estratti dai tessuti frequentemente perdono la loro attività dopo dialisi e che questa attività può essere riacquistata aggiungendo alle preparazioni enzimatiche quelle piccole molecole, i cofattori, che erano state rimosse durante la dialisi. Eppure nessuno aveva condotto ricerche sistematiche nel caso dell'emoglobina, benché sporadicamente fosse stato suggerito che ‛qualcosa' poteva essere stato perso con la dialisi. In effetti fin dal 1850 è noto che i composti fosforati del sangue sono prevalentemente contenuti negli eritrociti. Ora sappiamo che i fosfati organici hanno un ruolo cruciale nel controllare il trasporto dell'ossigeno. Nel 1967 R. e R. E. Benesch e, indipendentemente, A. Chanutin e R. R. Curnish hanno scoperto che il DPG, presente negli eritrociti umani in rapporto di circa una mole per mole di emoglobina, diminuisce grandemente l'affinità dell'emoglobina umana per l'ossigeno. Negli Uccelli e nelle tartarughe sembra che l'inositolo-esafosfato rimpiazzi il DPG nel ruolo di regolatore. In molti altri vertebrati inferiori questa funzione è svolta dall'adenosin-trifosfato (ATP).
L'emoglobina può essere correttamente considerata come una proteina che si lega con almeno quattro differenti leganti: O2, H+, CO2 e DPG. Il meccanismo con cui avvengono queste reazioni e le relazioni tra di esse si possono schematizzare come segue. I segmenti terminali della catena polipeptidica (sia dalla parte del gruppo -NH2 sia da quella del gruppo -COOH) hanno un ruolo fondamentale nel controllo della funzione dell'emoglobina. I quattro gruppi -COOH terminali possono ruotare liberamente nella ossiemoglobina, mentre nella desossiemoglobina la loro mobilità è ridotta da legami elettrostatici. Nella catena β della desossiemoglobina il gruppo imidazolico dell'istidina carbossiterminale è legato a un gruppo -COO- di un residuo di acido aspartico della stessa catena. Questo legame riduce la dissociazione del protone dall'imidazolo, e quindi la protonazione della desossiemoglobina è aumentata. Questo fatto rende conto di circa metà dell'effetto Bohr. Un altro ponte si costituisce tra le due catene α:

Questi ponti salini, presenti solo nella desossiemoglobina, riducono notevolmente la dissociazione dei gruppi NH3+ della valina rispetto a quella rilevabile nell'ossiemoglobina. Tale aumentato legame di protoni rende conto di circa un quinto dell'effetto Bohr alcalino. Questi gruppi possono anche, come dimostrato da Roughton, Kilmartin e Rossi-Bernardi, legare il CO2 mediante formazione di gruppi carbamminici, ma lo fanno solo nella forma non protonata:
−NH+3 ⇄ −NH2+H+
−NH2+CO2 &mis6;Q −NHCOO-+H+.
Queste reazioni avvengono anche a carico dei gruppi -NH2 terminali delle catene β, ma tali gruppi sono separati dai vicini -COOH terminali da una larga cavità: ed è in questa cavità che si lega il DPG in forma anionica. Il legame è molto forte, in quanto ai margini della cavità vi sono almeno 6 gruppi carichi positivamente. Questo è il sito più importante per il controllo del legame con l'ossigeno: in presenza di questo anione l'affinità dell'emoglobina per l'ossigeno diminuisce di dieci volte. In altre parole, la pressione d'ossigeno necessaria per raggiungere un certo grado di saturazione deve essere dieci volte maggiore. Bisogna notare inoltre che il DPG si lega tra i gruppi -NH2 terminali delle catene β solo se carichi e quindi compete con il legame del CO2:
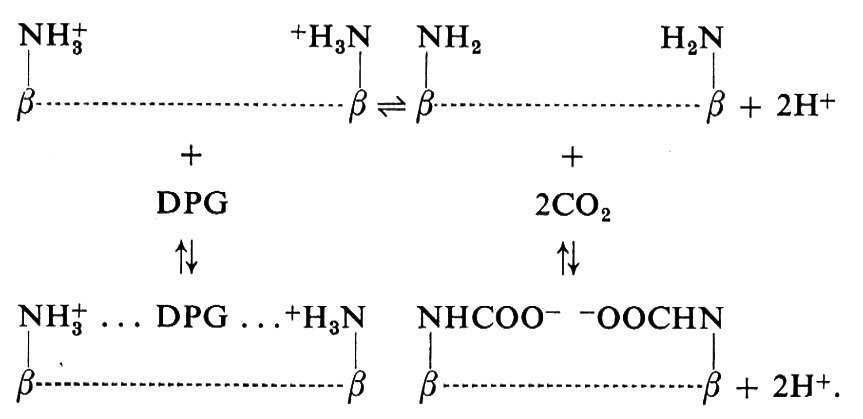
Sia il DPG che il CO2 sono legati essenzialmente dalla desossiemoglobina. Lo schema sopra riportato mostra come le relative proporzioni di legame di ciascuno di questi composti siano controllate dal pH. Quindi all'interno degli eritrociti l'affinità per l'ossigeno è controllata da un delicato equilibrio tra pH e concentrazioni di DPG e CO2.
4. Genetica ed evoluzione.
La determinazione della sequenza completa degli amminoacidi di un rilevante numero di emoglobine di Vertebrati e di molte altre proteine ha fornito una dovizia di nuove informazioni e di nuove idee sulla genetica e sull'evoluzione di queste proteine.
Gli emolisati di tutti i Vertebrati fin qui studiati contengono parecchie emoglobine differenti, spesso in concentrazioni ampiamente variabili. Nell'uomo adulto in aggiunta al componente predominante A vi sono due componenti minori, che rappresentano non oltre il 5%: sono le emoglobine A2 e A1c. Vi sono inoltre altri componenti, non ben caratterizzati, in minori proporzioni. L'emoglobina A2 contiene le medesime catene α, ma differenti catene, dette catene δ, al posto delle β. L'emoglobina A1c sembra identica alla comune emoglobina A, eccetto che vi è un esoso legato covalentemente ai gruppi -NH2 terminali delle catene β. È interessante notare che la concentrazione di questo componente è spesso grandemente aumentata in individui affetti da diabete mellito.
La maggior parte degli animali, ma non tutti, hanno emoglobine differenti durante la vita fetale o embrionale. Così, per esempio, il feto umano ha l'emoglobina F, la cui sintesi s'interrompe al momento della nascita; questa emoglobina contiene le stesse catene α dell'emoglobina A, ma le altre catene sono peculiari e vengono designate come γ. Tutte queste differenti catene polipeptidiche - α, β, γ e δ - si sono formate durante l'evoluzione per mezzo di duplicazioni di geni seguite da varie mutazioni, che hanno avuto come effetto numerose sostituzioni di amminoacidi. La dimostrazione del continuo verificarsi di queste mutazioni è fornita dalla presenza nel genere umano di più di 100 emoglobine ‛abnormi', in ciascuna delle quali si è verificata la sostituzione di un solo amminoacido.
Alcune di queste emoglobine anomale sono presenti con una certa frequenza: l'esempio più noto è l'emoglobina responsabile della malattia ereditaria detta anemia falciforme (v. sangue: Anemie emolitiche). La scoperta della sostituzione, in questa emoglobina, di un amminoacido, fatta da V. M. Ingram nel 1957, ha dato il via a innumerevoli ricerche, cosicché ora più di un migliaio di ricercatori in ogni parte del mondo scopre quasi ogni mese nuove emoglobine anomale grazie ad accurate indagini di massa. L'anemia falciforme è così chiamata in quanto la desossigenazione produce la trasformazione degli eritrociti dalla comune forma di disco biconcavo in fragili cellule a forma di falce, che si rompono facilmente causando di conseguenza gravi anemie. Inoltre, queste cellule ostacolano il flusso del sangue nei capillari tessutali. La malattia non curata è di solito fatale nello stato omozigote, mentre nello stato eterozigote sembra costituire addirittura un vantaggio: infatti gli individui eterozigoti sembrano meno colpiti dalla malaria. Il meccanismo di questo fatto rimane oscuro. La sostituzione di un amminoacido riguarda l'acido glutammico in posizione 6 della catena β rimpiazzato da una valina (tutte le posizioni sono numerate, per convenzione, a partire dal gruppo -NH2 terminale). Questa sostituzione fa sì che la desossiemoglobina formi una matrice gelificata semicristallina che distorce in qualche modo la forma della cellula. Il meccanismo con cui ciò avviene non è chiaro, ma si sa che la reazione del residuo di amminoacido amminoterminale con il cianato evita la gelificazione e ha grande valore terapeutico.
Le sequenze delle catene α e β dell'emoglobina umana sono riportate nella fig. 5, insieme con le sequenze della mioglobina di capodoglio e dell'emoglobina della lampreda di mare (Petromyzon marinus). Anche un superficiale esame di queste sequenze mostra diverse caratteristiche comuni. Molti studi comparativi di questo genere hanno dimostrato che tanto più sono vicini filogeneticamente gli animali studiati, tanto meno numerose sono le diversità nelle rispettive emoglobine. Per es., le catene α e β dell'emoglobina di gorilla differiscono per un solo amminoacido per catena da quelle dell'emoglobina umana. Se l'emoglobina di gorilla fosse stata presente in un uomo avrebbe potuto quasi certamente non essere scoperta per molto tempo.
Dettagliate comparazioni delle sequenze di molte emoglobine di Vertebrati hanno permesso la costruzione di un ‛albero filogenetico' basato sul numero di somiglianze (omologia) tra le diverse sequenze. Un ‛albero' di questo genere è riportato nella fig. 6 e indica i tempi di divergenza (cioè la duplicazione genetica) a partire da una forma ancestrale comune. Si trovano correlazioni relativamente buone tra l'ammontare dell'omologia e la sequenza evolutiva stabilita dai classici studi anatomici. Questi studi delle sequenze hanno portato alla conclusione che la velocità di sostituzione degli amminoacidi in una data proteina si è mantenuta relativamente costante nel tempo, cioè che il numero delle sostituzioni può essere considerato una specie di orologio biologico. Un corollario di questa osservazione è che la maggior parte del cammino evolutivo è avvenuto per mezzo di mutazioni ‛selettivamente neutre', cioè si ammette che il fissarsi delle mutazioni in una popolazione sia largamente indipendente dalla selezione naturale. Ma queste conclusioni sono inerenti a intervalli di tempo molto grandi; se fossero disponibili dati riguardanti intervalli di tempo relativamente brevi, la precisione dell'‛orologio delle mutazioni' sarebbe quasi sicuramente di gran lunga inferiore. Il suo funzionamento potrebbe essere paragonato all'andatura di un ubriaco che percorre barcollando una via: se egli compie abbastanza strada la precisione del suo cammino (deviazione dal centro della strada divisa per la distanza percorsa) potrà apparire elevata, mentre chi lo osserva da vicino ne trarrà una diversa impressione. In ogni caso la comparazione delle sequenze mostra quali parti siano mantenute e quali appaiano sostituibili facilmente, senza grandi ripercussioni. In generale, le sostituzioni di amminoacidi sulla superficie esterna dell'emoglobina tetramera hanno un'importanza fisiologica relativamente minore. Invece i residui idrofobici all'interno sono spesso conservati: l'inserzione di un residuo carico in un ambiente idrofobico può produrre gravi alterazioni. Studi sulla funzione di emoglobine umane anomale hanno mostrato che le sostituzioni nei punti di contatto delle subunità o in loro vicinanza hanno effetti rilevanti e spesso catastrofici.
Poiché questi punti di contatto sono indispensabili per la forma a S della curva di saturazione con l'ossigeno, le sostituzioni in queste zone spesso aboliscono la cooperatività, alterano grandemente l'affinità per l'ossigeno e possono perfino rendere non funzionante l'emoglobina. Anche i residui di amminoacidi con cui l'eme entra in contatto sono particolarmente importanti. La normale emoglobina A umana presenta una istidina su entrambi i lati dell'atomo di ferro: quella più vicina lega covalentemente il ferro (istidina prossimale); l'altra, che è più lontana, non lega direttamente il ferro (istidina distale). La molecola di ossigeno occupa lo spazio esistente tra il ferro e questa istidina distale. Sono state trovate emoglobine anomale aventi, al posto dell'istidina distale, una tirosina: ne risulta una situazione catastrofica dal punto di vista funzionale, in quanto si può formare un legame molto tenace tra il ferro trivalente e la tirosina:

L'equilibrio ossido-riduttivo
Fe2+⇄Fe3++e
risulta fortemente spostato verso destra e quindi viene rapidamente perduta ogni capacità di ossigenazione. Esistono molti esempi simili di rapporto tra struttura e funzione. Rapporti di questo genere aiutano a determinare quali siano i residui importanti funzionalmente e quali invece siano relativamente ‛accidentali'. Man mano che si scopriranno altre sostituzioni e se ne studierà la conseguenza fisiologica, si potrà ottenere una sempre più dettagliata conoscenza del funzionamento della molecola dell'emoglobina e della sua evoluzione.
Bibliografia.
Antonini, E., Brunori, M., Hemoglobin, in ‟Annual review of biochemistry", 1970, XXXIX, pp. 977-1042.
Dayhoff, M. O. (a cura di), Atlas of protein sequence and structure, Washington 1969, pp. D37-D65.
Dickerson, R. E., X-ray analysis and protein structure, in The proteins (a cura di H. Neurath), vol. II, London-New York 19642, pp. 603-778.
Eigen, M., Kinetics of reaction control and information transfer in enzymes and nucleic acids, in Nobel symposium V. Fast reactions and primary processes in chemical kinetics (a cura di S. Claesson), New York 1967, pp. 333-369.
Perutz, M. F., Stereochemistry of cooperative effects in haemoglobin, in ‟Nature", 1970, CCXXVIII, pp. 726-739.
Riggs, A. F., The haemoglobins, in The biology of lampreys (a cura di M. W. Hardisty e I. C. Potter), vol. II, London 1972, pp. 261-286.
Genetica del sangue
SOMMARIO: 1. Introduzione. □ 2. Genetica del globulo rosso: a) generalità sul globulo rosso; b) genetica dell'emoglobina; c) genetica degli enzimi del globulo rosso; d) genetica dei gruppi sanguigni. □ 3. Genetica del siero: a) generalità; b) le immunoglobuline (Ig), o anticorpi circolanti; c) il ruolo dell'antigene nella risposta immunitaria montata specificamente verso di esso; d) considerazioni di carattere generale sul sistema delle Ig. □ 4. Utilizzazione della genetica del sangue a fini teorici e applicativi: a) utilizzazioni teoriche; b) utilizzazioni pratiche. □ Bibliografia.
1. Introduzione.
Vi sono geni che si esprimono solo in cellule del sangue (o, più precisamente, negli organi emopoietici, come il midollo osseo) e i cui prodotti si trovano solo nel sangue. Gli esempi più noti sono i geni delle emoglobine e quelli della maggior parte dei gruppi sanguigni: è indubbio che la loro trattazione rientri a pieno diritto in questo articolo.
Esistono altri geni che, al contrario, funzionano solo in cellule che non hanno niente a che vedere con quelle del sangue e i cui prodotti non sono destinati a questo tessuto, come per esempio i geni per le funzioni specifiche del sistema nervoso. Anche per questi ultimi non vi sono dubbi: la loro trattazione non rientra negli scopi del presente articolo.
Ma per tutti gli altri geni è più o meno opinabile se debbano essere considerati come parte di questo capitolo della genetica. Pertanto, pur consapevoli che la nostra può essere considerata una decisione arbitraria, abbiamo ritenuto opportuno includervi anche i seguenti tre gruppi di geni: a) geni i cui prodotti - sebbene si formino al di fuori del sangue e degli organi emopoietici - sono destinati al sangue, anzi solo al sangue: fanno parte di questo gruppo i geni della sieroalbumina, delle aptoglobine e delle proteine della coagulazione, tutte proteine sintetizzate dal fegato; b) geni che funzionano in tutte le cellule (i geni cosiddetti house-keeping, perché servono ad assicurare le funzioni elementari comuni a tutte le cellule, indipendentemente dalle loro funzioni specifiche differenziative, quali un certo metabolismo di sostentamento, una certa sintesi proteica, la capacità di sfruttare l'energia chimica potenziale dell'ATP ecc.), ma di cui si conoscono variazioni le cui conseguenze fenotipiche si esplicano prevalentemente nel sangue: gli esempi più noti sono gli alleli Gd- del gene Gd della glucosio-6-fosfatodeidrogenasi (G-6-PD) - un tipico gene house-keeping -, alleli che si manifestano soprattutto nei globuli rossi; c) geni strutturali di enzimi anch'essi ubiquitari, o comunque molto diffusi, ma che di fatto si studiano solo nelle cellule del sangue, come i polimorfismi enzimatici ‛eritrocitari'.
Abbiamo invece tralasciato la trattazione di geni che, oltre a non essere esclusivi del sangue, si conoscono soprattutto per quadri clinici non ematologici a essi associati. Abbiamo ritenuto opportuno escludere anche quei geni che sono studiati nel sangue solo per ragioni di comodità tecnica (soprattutto facile disponibilità del materiale in esame), come molti geni dei cosiddetti ‛errori congeniti del metabolismo' che vengono studiati sui globuli rossi o sul siero, ma le cui conseguenze fisiopatologiche e cliniche riguardano prevalentemente, se non esclusivamente, altri tessuti.
Che cosa rimane della genetica umana in questo articolo dopo avere operato queste esclusioni? È molto facile rispondere: ‛quasi tutto' se si considera non tanto la quantità (cioè il numero di geni) quanto la qualità, cioè la loro importanza concettuale per la genetica umana (basti pensare che fanno parte della genetica del sangue i gruppi sanguigni, le emoglobine e le immunoglobuline); ‛molto poco' se invece si tiene conto del numero dei geni inclusi. Infatti se si guarda l'elenco dei circa duemila marcatori genetici unifattoriali finora individuati nell'uomo, pubblicato da McKusick, si constata che solo una minoranza di essi sono ‛geni del sangue'. Però è proprio questa minoranza il vero nucleo della genetica umana (v. gene; v. genetica: Genetica).
2. Genetica del globulo rosso.
a) Generalità sul globulo rosso.
Il globulo rosso, o eritrocita, è la cellula deputata al trasporto dell'ossigeno ai tessuti e dell'anidride carbonica in senso inverso. Entrambe queste funzioni sono effettuate senza alcun dispendio di energia dall'emoglobina (Hb) e tutte le altre attività eritrocitarie sono a esse subordinate.
Il globulo rosso è costituito quindi di emoglobina e di tutto ciò che serve per farla funzionare. Che non è poco: c'è una bella differenza funzionale tra trecento milioni di molecole di emoglobina libera in soluzione e i trecento milioni di molecole di emoglobina di un globulo rosso; queste ultime infatti non solo rimangono funzionalmente efficienti per tutta la durata di vita dell'eritrocita, ma si trovano in un ambiente biochimico peculiare che ne ottimizza l'efficienza media ed è anche specificamente in grado di regolame il funzionamento.
Il compito del ‛resto' del globulo rosso consiste allora: 1) nel creare l'ambiente biochimico adatto al funzionamento dell'Hb e nel mantenerlo sostanzialmente inalterato per circa 120 giorni, e a questo provvede il metabolismo del globulo rosso; 2) nel delimitare questo ambiente particolare controllandone gli scambi con l'esterno, e a questo provvede l'involucro del globulo rosso (o membrana eritrocitaria).
L'economia con cui l'eritrocita svolge queste sue funzioni ‛accessorie' è sorprendente: dato che l'Hb costituisce il 95% del suo peso secco, è solo al restante 5% che spetta il compito di costituire una membrana, effettuare tutte le funzioni richieste per la sopravvivenza dell'intera cellula e assicurare che l'Hb continui con piena efficienza il trasporto dell'O2 e del CO2. Incapace di rinnovare il suo patrimonio di proteine strutturali ed enzimatiche e molto a corto di energia (nell'eritrocita, privo com'è di organelli, non si effettua né sintesi proteica né fosforilazione ossidativa), questa cellula, che è stata definita (Valentine, 1979) ‟una cellula nomade che, sul piano metabolico, parte con poco e muore con ancora meno", riesce, fra l'altro, per ben quattro mesi: 1) a mantenere la sua forma peculiare di disco biconcavo e l'integrità, deformabilità ed elasticità della sua membrana, malgrado le intense e frequentissime sollecitazioni meccaniche a cui è sottoposta nei vortici cardiaci e quando si fa strada all'interno di capillari, come quelli splenici, di diametro molto inferiore a quello eritrocitario; 2) a pompare cationi contro gradienti elettrochimici; 3) a sintetizzare glutatione e sostanze, come il 2,3-difosfoglicerato (2,3-DPG), specificamente destinate a influenzare il comportamento funzionale dell'Hb; 4) a proteggere l'Hb contro la sua trasformazione in metemoglobina e a impedire, inoltre, la denaturazione ossidativa non solo dell'Hb, ma anche di molti altri componenti cellulari, tra cui i lipidi della membrana; 5) a prevenire non solo la fusione, ma spesso anche il semplice contatto con altri eritrociti, che la priverebbe di una parte della superficie di scambio essenziale per la sua funzione di trasporto.
Tutto questo non basterebbe a spiegare come mai il globulo rosso sia stato e continui a essere la ‛cellula eucariotica' (se cosi si può chiamare, pur essendo, nei Mammiferi, priva di nucleo) di gran lunga più studiata e meglio conosciuta ai livelli metabolico, molecolare (Hb) e sopramolecolare (membrana), dato che non c'è alcun motivo di credere che l'evoluzione l'abbia resa più adatta delle altre a svolgere la sua funzione. Si può al contrario dare per scontato che di regola le cellule, di qualsiasi tipo esse siano, sono fatte su misura per il compito a cui sono destinate, ma la loro specificità è molto più difficile da documentare.
Il globulo rosso, infatti, è uno di quei materiali biologici, come la drosofila, la cellula Escherichia coli, alcuni batteriofagi, l'uovo di riccio di mare e pochi altri, che sembrano creati apposta per stimolare i biologi ad affrontare certi problemi e metterli in grado di risolverli. E questo per motivi sia tecnici sia concettuali: per esempio, è un materiale bioptico estremamente abbondante e accessibile; è una cellula molto semplice, ma non tanto da essere considerata poco interessante; contiene in grandissima quantità e quasi allo stato puro una proteina, l'Hb; essendo priva di organelli, contiene un solo tipo di membrana, quella cellulare, come i Batteri, che però sono procarioti; è facilmente agglutinabile da anticorpi diretti verso alcuni suoi antigeni di membrana, la cui variabilità genetica si è prestata a essere studiata con la tecnica particolarmente semplice dell'agglutinazione.
Le caratteristiche eritrocitarie più note perché comunemente utilizzate a livello clinico, essendo apprezzabili con una strumentazione estremamente semplice, sono presentate nella tab. I (v. anche cellula: Fisiologia; v. sangue: Emoglobina e Organi emopoietici).

b) Genetica dell'emoglobina.
La maggior parte delle acquisizioni sulla genetica dell'Hb, che ha rappresentato fin dal suo nascere una delle frontiere più avanzate della biologia molecolare, costituisce un complesso di conoscenze che, sebbene tutt'altro che antiche, sono ormai classiche e perfettamente chiare; ciò è soprattutto vero per ciò che riguarda la struttura e il funzionamento dei geni strutturali, su cui ci soffermeremo relativamente poco per poter dedicare maggiore attenzione agli aspetti che rientrano tuttora tra i problemi maggiori della biologia.
1. Requisiti funzionali del sistema dell'Hb. - Il corretto andamento del trasporto dell'ossigeno, che è il compito dell'Hb, è assicurato solo se sono soddisfatte tre serie di esigenze: strutturali, quantitative e ontogenetiche.
È dalla struttura delle catene globiniche che dipendono tutte le proprietà funzionali dell'Hb (v. sangue: Anemie emolitiche ed Emoglobina); ciò significa non solo che la molecola deve rimanere stabile nel suo complesso, ma che nessuna delle sue caratteristiche deve alterarsi durante i circa 120 giorni che costituiscono la durata media di permanenza in circolo di un globulo rosso normale. Tra le 300 e più varianti genetiche dell'Hb finora note (più di 150 della catena β, più di 100 della α, circa 25 tra γ e δ, più altre da fusioni di geni diversi), alcune non sono associate ad anomalie funzionali evidenti; altre, invece, comportano alterazioni di una o più proprietà, quali anomalie concernenti l'affinità dell'Hb per l'O2, l'effetto Bohr, la sensibilità al 2,3-DPG, la stabilità, l'affinità tra catene α e catene β, la tendenza a ossidarsi dell'atomo di Fe2+ dell'eme (metemoglobine), e cosi via. Alcune di esse, in particolare le varianti molto instabili che, precipitando all'interno dell'eritrocita, ne alterano la struttura compromettendone la vitalità e causando quindi un'anemia emolitica talora molto grave, rappresentano esempi molto illustrativi di mutazioni letali o sub-letali dominanti: l'allele patologico non si limita a essere silente (e in questo caso - che è di gran lunga il più comune - poiché la funzione di un solo allele normale è sufficiente ad assicurare un buono stato di salute, l'allele patologico è recessivo: la funzione necessaria manca solo se l'allele normale è assente, cioè se l'allele non funzionante è allo stato omozigote), ma produce qualcosa di intrinsecamente dannoso, donde la dominanza.
Alcuni dei valori quantitativi del sangue normale sono assoluti: esso contiene una certa concentrazione di molecole di Hb pari a 1,5•1018 molecole per centimetro cubo, o ml (3•108 molecole per eritrocita × 5•109 eritrociti per ml); altri, di gran lunga più interessanti dal punto di vista genetico, sono invece relativi, in quanto tutte le Hb normali sono tetrameri costituiti da un dimero di catene di tipo α e da un dimero di catene non-α, per cui normalmente le sintesi di questi due tipi di catene si equivalgono in tutti i periodi della vita (v. fig. 1A). Quando questo equilibrio sintetico non è rispettato, e ciò di regola è dovuto al fatto che la sintesi delle catene di un certo tipo è ridotta se non addirittura assente, si parla di talassemia (o microcitemia) α o β, a seconda che sia compromessa la sintesi delle catene α o di quelle β (v. sangue: Anemie emolitiche).
Le esigenze ontogenetiche sono sovrapposte a quelle precedenti: in tutti i periodi dello sviluppo le Hb funzionano normalmente solo se sono costituite da un dimero di catene di tipo α e da un dimero non-α che assicurino loro le proprietà funzionali fondamentali necessarie in tutte le condizioni fisiologiche; ma è molto importante che le Hb abbiano qualcosa di più, che cioè siano adatte al trasporto dell'O2 nelle condizioni in cui esso deve effettivamente essere espletato. Non è la stessa cosa avere come fonte di O2 l'atmosfera gassosa degli alveoli polmonari o l'HbO2 situata dentro i globuli rossi del sangue materno, e in quest'ultimo caso è verosimile che faccia differenza se lo scambio si svolge nelle lacune di Wolff del sacco vitellino dell'embrione o nella placenta del periodo fetale. È inoltre presumibile che nei periodi embrionale, fetale ed extrauterino (o ‛adulto', come impropriamente viene di solito chiamato) esistano altre differenze rilevanti per la funzione del trasporto dell'O2 e del CO2. A queste condizioni fisiologiche diverse fa riscontro l'esistenza di Hb diverse in periodi diversi dello sviluppo (v. fig. 1B e tab. II). All'inizio c'è l'Hb embrionale (ζ2ε2) cui segue l'Hb fetale o Hb F (α2γ2); compaiono infine le Hb adulte (Hb A: α2β2, che costituisce circa il 97-98% dell'Hb della vita extrauterina, e Hb A2: α2δ2, che costituisce, insieme a tracce persistenti di Hb F, il restante 2-3% dell'Hb del periodo ‛adulto'). Nei periodi di transizione catene globiniche ed emoglobine caratteristiche di periodi diversi coesistono nell'organismo (per es. Hb F e Hb A) o anche entro le stesse molecole (per es. ζ2γ2; α2ε2; v. tab. II). Si conoscono anche per questo aspetto del funzionamento del sistema delle Hb alterazioni genetiche, chiamate HPFH (Hereditary Persistence of Fetal Haemoglobin), nelle quali la sintesi dell'Hb F continua per tutta la vita, talora in quantità molto elevate (nella forma negra allo stato omozigote tutta l'Hb è di tipo fetale).


Da un punto di vista genetico, le nostre conoscenze sulla Hb (v. tab. III) variano grandemente: sappiamo tutto sulla sua struttura, molto sulla regolazione quantitativa e qualcosa sulla regolazione ontogenetica della sua sintesi; nulla sappiamo invece sul controllo del tipo di cellule e di organi in cui questi geni normalmente si esprimono e sarebbe comunque estremamente difficile scoprire alterazioni di questo tipo di controllo, ammesso che esistano.
2. Struttura e funzionamento dei due clusters globinici. - La struttura del cluster α e del non-α, presentata in forma schematica nella fig. 1, è nota in considerevole dettaglio (v. fig. 2), nel senso che sono stati individuati e localizzati i geni strutturali che ne fanno parte, misurate con buona approssimazione le distanze che li separano e, soprattutto, determinate le sequenze complete delle regioni codificanti di quasi tutti questi geni e anche di notevoli tratti delle loro regioni non codificanti. Questi ultimi dati hanno permesso di analizzare con una precisione nemmeno sperabile fino a pochi anni fa gli aspetti anatomo-funzionali di questi geni, cioè di descriverne con assoluta completezza la struttura, comprendendone allo stesso tempo non solo la funzione globale, ma come essa risulti dalla cooperazione delle sue varie parti, di molte delle quali sono stati identificati il compito e il significato precisi (v. fig. 3).
La fig. 4 presenta molto schematicamente i tre processi fondamentali dell'espressione di un gene strutturale (è preso come esempio un gene globinico): la trascrizione (DNA → pre-mRNA), la maturazione (pre-mRNA → mRNA) e la traduzione (mRNA → proteina) (v. acidi nucleici; V. biologia; v. gene).
3. Concetti di portata biologica generale a cui si è giunti attraverso lo studio delle emoglobine. - α) Nascita della patologia, o meglio, della genetica molecolare. La scoperta da parte di Pauling, Itano e Singer, nel 1949, che un carattere mendeliano (globuli rossi di forma normale oppure a falce) dipende dal tipo di Hb presente (Hb A oppure Hb S) costituisce una pietra miliare della biologia, paragonabile al passaggio dall'anatomia all'istologia e alla citologia. Anche in questo caso si è trattato di un cambiamento di livello: dalla cellula alla molecola, dato che per la prima volta si è descritto un fenotipo a livello molecolare. Il fatto che due alleli producessero due proteine alleliche costituiva la prova diretta che la conformazione di una proteina dipende dalla conformazione del gene per quella proteina. Questa scoperta è una delle basi su cui poggia il celebre concetto ‛un gene - una proteina', da cui si sono poi sviluppate la genetica biochimica e quella molecolare (v. biologia).
Sempre in questa direzione, poco meno che dieci anni più tardi è giunta, a opera di Ingram, un'altra scoperta, anch'essa di portata storica: l'Hb S differisce dall'Hb A per una singola sostituzione amminoacidica, che è come dire che ogni gene (strutturale, come lo chiamiamo ora) determina la struttura della catena polipeptidica corrispondente, amminoacido per amminoacido. Dato che nel frattempo si era delucidata la struttura del DNA e se ne era scoperto il significato di sostanza depositaria delle proprietà genetiche, questa era l'unica tessera mancante per poter formulare la domanda: ‟Come fa una sequenza di desossiribonucleotidi a determinare univocamente una sequenza amminoacidica?" In altri termini, era nato il problema del codice genetico, che verrà decifrato definitivamente nel 1966.
Nel 1961 è stata poi determinata la sequenza completa delle catene α e β dell'Hb umana e, nel breve volgere di pochi anni, quella di altre catene globiniche dell'uomo e di altre specie; veniva così dimostrata la comune origine da un gene ancestrale di tutti questi geni strutturali. Nasceva in tal modo la filogenesi a livello molecolare proteico; in questi ultimi anni, grazie alle determinazioni - succedutesi a ritmo letteralmente vertiginoso - di nuove sequenze nucleotidiche, la filogenesi sta passando anche al livello del gene, direttamente.
Sempre all'inizio degli anni sessanta, la determinazione delle altre strutture della molecola emoglobinica ha permesso di identificare la sua architettura sterica ben più dettagliatamente che per qualsiasi altra molecola proteica. In seguito, si è arrivati a conoscere le strutture secondaria e terziaria di molte altre proteine, ma l'Hb è tuttora la sola in cui è stato possibile verificare le ipotesi anatomo-funzionali sul ruolo delle varie parti della molecola nel determinismo delle sue proprietà globali. Ci si è potuti valere, infatti, delle centinaia di varianti emoglobiniche note, per molte delle quali è stato possibile correlare una precisa alterazione strutturale (quasi sempre una singola sostituzione amminoacidica) con alterazioni steriche e funzionali perfettamente identificate. È la ben nota storia della patologia che viene in aiuto del fisiologo compiendo per lui esperimenti tecnicamente inattuabili: in questo caso si trattava di un'alterazione naturale riguardante un segmento molto corto e precisamente identificato di una molecola.
β) I clusters. Il concetto che geni con funzioni simili tendono a stare vicini, cioè a formare un cluster, non è certo entrato nella biologia per la porta dell'Hb, anzi nemmeno attraverso quella, molto più ampia, degli Eucarioti: l'operone di Jacob e Monod è infatti un'entità tipicamente batterica. Ma sistemi come quello dell'Hb, delle immunoglobuline e dell'HLA, con i relativi geni responsabili delle funzioni effettrici della risposta immunitaria o almeno con esse correlati, hanno contribuito, ciascuno con le proprie connotazioni, a immettere tale concetto nel philum dei Mammiferi.
Per restare nel campo dell'Hb, tutti i geni adibiti alla sintesi delle catene di tipo α sono addensati nell'esiguo spazio di circa 25 Kb (Kb = Kilobase, cioè mille basi o coppie di basi) nel cromosoma 16, e tutti quelli delle catene non-α in uno spazio quasi altrettanto esiguo, circa 50 Kb, che costituisce circa un tremillesimo del cromosoma 11. Non solo, ma i geni che devono sintetizzare globine dello stesso tipo, nello stesso periodo dello sviluppo, sono ancora più addensati gli uni agli altri, tanto da formare dei sotto-clusters, i cosiddetti blocchi ontogenetici, i quali si spengono e si accendono in modo mutualmente esclusivo seguendo un'unica direzione che - non sappiamo se per caso o per qualche ragione - coincide con quella in cui si svolge la trascrizione. Infine tutti questi geni sono trascritti dallo stesso filamento di DNA.
Anche se non sappiamo ancora i motivi di queste precise relazioni spaziali, di una cosa si può comunque essere certi: esse sono indispensabili alla corretta regolazione di questi geni (v. tab. III), come si può dedurre dal fatto che alterazioni della loro posizione risultano in anormalità del loro comportamento quantitativo e/o ontogenetico.
γ) Coordinazione delle sintesi dell'eme, delle catene di tipo α e di quelle non-α. Ogni globulo rosso umano normale contiene circa trecento milioni di molecole eterodimeriche di Hb, la cui formula generale è: (catena di tipo α)2 (catena di tipo non-α)2, mentre non vi si trovano quantità apprezzabili di omodimeri del tipo β4 o γ4, né catene o dimeri liberi, né eme. Questo significa che (escludendo la possibilità che catene globiniche vengano distrutte prima di venire a far parte di una molecola di Hb completa, fenomeno praticamente inesistente in condizioni normali) i geni emoglobinici sintetizzano durante la maturazione dell'eritroblasto un numero di catene di tipo α pari a quello delle catene non-α e pari anche al numero di molecole di eme (v. figg. I e 4); infatti tutte queste catene si trovano alla fine a far parte delle tipiche molecole tetrameriche di Hb.
Per raggiungere questo risultato così preciso non basterebbe nemmeno che queste tre attività sintetiche semplicemente si equivalessero: questa è una condizione necessaria, ma non sufficiente. Occorre anche che siano coordinate nel tempo, dato che le catene globiniche, soprattutto le α, sono molto instabili se non sono associate alle loro controparti in un eterodimero, e in assenza dell'eme precipiterebbero in brevissimo tempo. In altre parole, le catene globiniche non possono aspettare: esse devono essere sintetizzate nella quantità giusta e al momento giusto, che è quello in cui vengono sintetizzate le altre.
Le nostre conoscenze su come venga raggiunto questo perfetto equilibrio quantitativo e temporale sono molto scarse. Si sa però che l'inizio della traduzione degli mRNA per le globine è di regola bloccato da un inibitore e che questa inibizione può essere rimossa dall'eme, che può quindi dare via libera alla traduzione. Inoltre la traduzione delle catene β è stimolata dalle catene α, che favoriscono il rilascio delle catene β ormai complete dal complesso βmRNA-poliribosoma-tRNA-catena β (v. acidi nucleici; v. biologia; v. genetica: Citogenetica). Non si sa quale sia il ruolo esatto di questi meccanismi, anche se è presumibile che abbia grande importanza; infatti il primo di essi determina il blocco della sintesi globinica in assenza dell'eme e il secondo fa sì che le catene α libere si procurino le β a cui associarsi favorendone la sintesi. A ogni modo, il bilancio sintetico α/non-α è ottenuto con meccanismi completamente diversi da quello che opera per l'insulina; quest'ultima, infatti, è costituita da due catene, A e B, che derivano da un'unica catena ACB, la proinsulina, per rimozione del tratto intercalare C, per cui le catene A e B si generano necessariamente in egual numero.
Si conosce tutta una serie di condizioni, le talassemie o microcitemie, nelle quali, essendo depressa o addirittura abolita la sintesi delle catene α (α-talassemie) oppure quella delle β (β-talassemie), è compromesso più o meno gravemente il bilancio sintetico α/non-α. Questo rapporto diventa infatti minore di 1 nelle α-talassemie e maggiore di 1 in quelle β, e le catene globiniche che restano libere tendono a precipitare nel corso della maturazione dell'eritroblasto, che viene spesso interrotta (eritropoiesi abortiva o inefficace). L'entità di questi disturbi dipende da una serie di fattori, primo fra tutti il genotipo del paziente: se è eterozigote, cioè se uno dei due clusters funziona normalmente (anzi, spesso ancora più del normale, con meccanismo compensatorio), allora il suo stato di salute è normale o appena alterato (talassemia minima); se invece nessuno dei suoi due clusters α o dei suoi due clusters non-α ha un'attività sintetica paragonabile a quella normale, ne risulta un quadro clinico gravissimo che, nel caso della β-talassemia, prende il nome di morbo di Cooley (v. sangue: Anemie emolitiche).
Le talassemie hanno grande interesse da un punto di vista epidemiologico dato che sono largamente diffuse in diverse aree molto popolate della Terra. Solo in Italia, per esempio, si stima che i portatori eterozigoti siano circa due milioni e che nasca ogni giorno un bambino destinato a essere affetto da morbo di Cooley (alla nascita la sintomatologia non è ancora manifesta, perché la maggior parte delle catene non-α è ancora di tipo γ, ed è pertanto prodotta da geni che sono normali in questi soggetti). Attualmente si sta cercando di mettere a punto procedimenti di diagnosi prenatale che potrebbero permettere la prevenzione, mediante aborto selettivo, della nascita di un certo numero di soggetti destinati ad ammalarsi di morbo di Cooley. È chiaro tuttavia che una prevenzione efficace può essere effettuata solo dopo che un preventivo censimento abbia messo in luce quali siano le coppie che corrono il rischio di generare figli affetti da morbo di Cooley.
In campo biologico l'interesse teorico delle talassemie è almeno duplice: per la biologia molecolare e dal punto di vista evoluzionistico, in quanto esempi di polimorfismi ‛malarici'. Che le talassemie siano da sempre uno degli argomenti preferiti della biologia molecolare più avanzata lo si spiega non tanto con il fatto che costituiscono esempi ben documentati di compromissione della funzione di un gene strutturale, quanto perché per la loro estrema eterogeneità esse offrono un assortimento ricchissimo dei vari tipi di malfunzionamento di un gene strutturale; anzi, perseguendo sempre più in dettaglio il loro studio a livello di DNA, ci si attende di trovare alterazioni molecolari di nuovo tipo che gettino luce su modalità della sintesi proteica ancora ignote e permettano di identificare regioni e particolarità dei tratti di DNA adiacenti ai geni strutturali, che siano rilevanti per la loro espressione.
Come si è detto, l'individuazione della base molecolare delle talassemie si è rivelata un vero e proprio vaso di Pandora: sono stati individuati casi in cui l'assenza di produzione di una determinata globina era dovuta all'assenza dei geni strutturali incaricati di produrla (è questo il motivo più ovvio per un'assenza di funzione; tuttavia la sua scoperta, avvenuta nel 1974, ha rappresentato il primo caso in cui si è documentata negli Eucarioti la base molecolare di un certo fenotipo direttamente a livello di DNA); in altri casi la compromissione più o meno completa della sintesi di una catena era dovuta a un cambiamento del codone term che non funzionava più da segnale di arresto della traduzione; in altri ancora, un codone che normalmente specifica un amminoacido era diventato una tripletta d'arresto di traduzione; oppure si era verificata la delezione di un grosso tratto 3′ del gene; fino a un caso in cui la mutazione responsabile aveva creato un punto alternativo di splicing di un introne del βpre-mRNA, per cui da una famiglia di molecole tutte uguali di questo pre-mRNA si originavano due tipi di βmRNA, di cui uno, quello normale, derivava da una molecola maturata in modo normale, mentre l'altro tipo di molecola risultava intraducibile. E l'elenco potrebbe ancora proseguire.
Il riassumere in modo schematico i progressi compiuti in questo campo (v. fig. 5) ha un interesse non solo storico, ma anche predittivo. Infatti, poiché per gli Eucarioti siamo pervenuti a una fase così avanzata della comprensione di una classe di disturbi regolativi solo nel caso della sintesi dell'Hb, il ricapitolarne le fasi principali è un modo per prevedere quali saranno le tappe che dovremo superare nel chiarimento dei disturbi regolativi di altri sistemi genetici.
Per chiarire la ezio-patogenesi delle talassemie, se restiamo fedeli alla rappresentazione schematica illustrata nella fig. 5, dobbiamo riuscire a spezzettare le frecce lunghe in frecce sempre più corte (da ----→ a --→ --→ a → → → ecc.). Il lavoro può dirsi completo per una determinata forma di talassemia solo quando, oltre ad avere individuato l'alterazione del DNA che ne è la causa, si sono identificate anche, una per una, tutte le singole tappe patogenetiche che conducono da quella alterazione del DNA al fenotipo talassemico corrispondente.
Nel passaggio dalla 2a alla 3a fase (quella attuale) ci si è trovati di fronte a un risultato del tutto inatteso, e cioè l'esistenza di molteplici forme sia di α- sia di β-talassemie. Nonostante le controversie che tale risultato ha determinato sul finire degli anni sessanta, il concetto che le talassemie costituiscono un'entità estremamente eterogenea da moltissimi punti di vista (tipo di sintesi compromesse: α o β; tipo di alterazione genetica; tipo di meccanismo biosintetico anormale: trascrizione, maturazione, traduzione; ecc.) è definitivamente acquisito, grazie soprattutto all'efficienza delle tecniche della biologia molecolare.
Nel periodo in cui l'estrema eterogeneità delle talassemie veniva ancora accettata con riluttanza, nessuno metteva ormai più in discussione il loro significato evolutivo: si tratta di polimorfismi che conferiscono resistenza alla malaria perniciosa. Una volta accettato che il fenotipo talassemico con la conseguente resistenza alla malaria insorge qualunque sia il modo in cui si instaura uno sbilancio sintetico α/non-α, non si vede perché tutte le popolazioni esposte a questo agente selettivo dovrebbero necessariamente avere in comune lo stesso allele talassemico, tra i numerosissimi che, ciascuna per conto suo, avrebbero potuto produrre per mutazione, o in qualsiasi altro modo, e poi selezionare. Quindi se questo problema fosse stato preso in esame da biologi molecolari insieme a studiosi di evoluzione, e non per compartimenti stagni, ci si sarebbe dovuto aspettare - salvo a confermarlo poi sperimentalmente - quello che si è poi in effetti constatato, cioè che ogni popolazione esposta per tempi evolutivamente significativi a un'intensa endemia malarica, si era costruita i suoi propri alleli talassemici. Del resto, avrebbe dovuto costituire un buon indizio in questo senso il fatto che, al livello di analisi utilizzabile allora, quello proteico, l'eterogeneità era stata già dimostrata: esistevano α- e β-talassemie, a loro volta comprendenti rispettivamente la α-Constant-Spring e altre α-talassemie diverse, e le talassemie Lepore e le δβ o F-talassemie, e altre ancora. Non si vede perché l'eterogeneità avrebbe dovuto limitarsi al solo livello raggiungibile con le tecniche di quel periodo.
La diffusa sensazione che ci fossero pochissime forme di talassemia α e pochissime di non-α è stata probabilmente determinata dalla lentezza con cui i biologi hanno preso atto che la variabilità genetica è in generale molto grande: ormai si può dire che qualunque fenotipo, sia normale sia patologico, pur apparentemente omogeneo a livello fenotipico, quando è stato analizzato abbastanza a fondo si è dimostrato più o meno eterogeneo. Quindi occorrerebbe ormai desistere da un atteggiamente troppo rigoroso quando si tratta di accettare prove di eterogeneità e troppo accomodante quando si tratta di accettare l'ipotesi di una omogeneità che talvolta viene data addirittura per scontata. Anzi, che in mancanza di dati si dovrebbe se mai propendere per una eterogeneità genotipica, vale soprattutto nel caso di alleli patologici che, almeno in una combinazione genotipica, conferiscono un fenotipo chiaramente svantaggioso. Infatti, se sono rari (per esempio, ogni singolo tipo di errore congenito del metabolismo dovuto a un deficit enzimatico specifico) sono frutto diretto della mutazione, cioè di eventi mutazionali in genere diversi tra loro - ci sono infiniti modi di sovvertire una struttura complessa e organizzata come un gene - che, verificandosi a caso, alterano in un modo o nell'altro il gene dell'enzima in questione; e se sono comuni, come nel caso della talassemia e della enzimopenia per la G-6-PD, sono lo stesso quasi sempre eterogenei almeno a livello interpopolazioni (ma spesso anche intrapopolazioni) per convergenza evolutiva (v. razza).
δ) La clonogenesi ricapitola l'ontogenesi. Più precisamente: l'ontogenesi di ogni clone ricapitola quella dell'intero organismo. Abbiamo già descritto in precedenza i vari switches che si succedono durante lo sviluppo nell'espressione dei blocchi ontogenetici sia α sia non-α; i meccanismi fini che presiedono a questi fenomeni sono per ora ben lontani dall'essere compresi, ma alcuni fatti importanti sono ormai acquisiti.
Anzitutto, come si era previsto, è al livello della trascrizione che si regola l'entrata in azione sequenziale di questi geni: se non è ancora il momento che certi geni globinici producano globine, essi non vengono trascritti. Sarebbe stato veramente strano che tutti i geni globinici fossero trascritti, ma solo certi mRNA fossero tradotti.
Il comportamento ontogenetico di questi geni non dipende dalla loro struttura, ma da quella del cluster in cui si trovano: geni strutturali assolutamente normali funzionano in periodi ‛sbagliati' se si trovano in clusters anormali. Per esempio, i geni γ dei clusters con HPFH funzionano durante tutta la vita extrauterina con altissima efficienza (tanto da vicariare quasi perfettamente la funzione del gene β che è assente) sebbene siano del tutto normali, a causa di un'estesa delezione alla loro destra che comprende l'intero blocco adulto e parte della regione normalmente interposta tra questo e il blocco fetale (v. fig. 2). Quando invece la delezione della parte destra del cluster non-α è meno estesa, tanto da lasciare la parte sinistra del gene δ (v. fig. 2), i geni γ sono solo parzialmente derepressi, cioè producono catene γ in quantità considerevoli, ma non abbastanza da prevenire l'insorgenza di un quadro talassemico (δβ o F-talassemie): si ha allora un quadro fenotipico intermedio tra la talassemia β, in cui la sintesi delle catene β è compromessa e i geni γ non sono affatto derepressi (questa è la forma più grave e allo stato omozigote corrisponde al morbo di Cooley), e la HPFH, in cui la sintesi delle catene β è abolita (il gene β è addirittura mancante) ma il perfetto funzionamento dei geni γ previene qualunque conseguenza patologica, anche negli omozigoti. Si tratta, come si vede, di tipici effetti di posizione cis-dominanti (cis, perché limitati ai geni γ che si trovano in cis rispetto alla delezione; dominanti, perché questi geni funzionano in modo anormale anche negli eterozigoti per un cluster non-α del tutto normale). Si conoscono anche delle forme di HPFH dovute a mutazioni di geni indipendenti che, evidentemente, controllano il funzionamento ontogenetico dei geni globinici con fattori diffusibili e non presentano quindi effetti di posizione.
Una delle linee di ricerca più promettenti e affascinanti sulla regolazione ontogenetica dei geni emoglobinici è certamente quella del suo studio su cellule in coltura. È stato così possibile stabilire in che cosa consista il ben noto switch feto-adulto a livello di organismo; tale switch, infatti, non fa sì che i doni eritropoietici dopo la nascita siano comunque destinati a produrre catene β (+δ) e a non produrre catene γ: questo accade solo se l'eritropoiesi si svolge con ritmo normale; se invece, per qualsiasi motivo, essa si svolge in modo molto accelerato o addirittura tumultuoso, come per esempio nel morbo di Cooley, doni geneticamente normali producono catene γ. In altre parole, se cellule ormai impegnate in senso eritropoietico, ma solo potenzialmente (non stanno, cioè, ancora sintetizzando Hb), vengono reclutate da un clone ancora giovane, che ha effettuato poche moltiplicazioni cellulari, per essere avviate nell'ultimo tratto della differenziazione, ossia alla sintesi dell'Hb, il risultato è che, anche se appartengono a un soggetto adulto, producono Hb F, cioè esprimono i loro geni γ. Ciò non ammette che una interpretazione. Le prime cellule di ogni clone entropoietico di un soggetto adulto sono predisposte a trascrivere i loro geni γ (e forse le primissime cellule sono predisposte addirittura a trascrivere il gene ε), e questo effettivamente si verificherebbe se esse venissero reclutate a questo stadio. Di regola, invece, questo non accade, e il clone può cosi continuare a moltiplicarsi; durante questo ulteriore periodo le sue cellule completano il switch feto-adulto, per cui, quando finalmente imboccano la via terminale della differenziazione, diventando eritroblasti, e trascrivono i geni non-α predisposti per la trascrizione, hanno ormai fatto in tempo a rendere non trascrivibili i geni γ e trascrivibili i geni δ e β.
Esistono quindi due fasi. Nella prima, quella della moltiplicazione di cellule ormai potenzialmente - ma solo potenzialmente - eritropoietiche, le cellule si preparano alla loro futura attività e anzitutto rendono i geni embrionali non trascrivibili, neppure potenzialmente, e, viceversa, potenzialmente trascrivibili quelli fetali; poi, se non sono reclutate, hanno ancora tempo per effettuare il secondo switch, che in condizioni normali completano prima di avviarsi a diventare eritroblasti. Nella seconda fase le cellule semplicemente esprimono i geni predisposti in tal senso nella fase precedente.
In conclusione, ogni clone di un individuo adulto normale è prima embrionale, poi fetale e infine adulto. Si esprimono tutti come adulti non per una loro intrinseca proprietà, ma perché hanno avuto tutto il tempo, ciascuno per conto proprio, di ‛ricapitolare' l'ontogenesi passando attraverso tutte le sue fasi.
Sull'esatta attività delle cellule di ogni clone pre-eritropoietico durante il tempo necessario per effettuare questi switches si sa ancora molto poco. Si è qui nel vero cuore della differenziazione ontogenetica, e la comprensione degli eventi che la caratterizzano significherebbe il chiarimento di molti dei fenomeni ancora più o meno misteriosi dello sviluppo. Non si tratta di un compito facile, ma in compenso lo si affronta sulla regione meglio conosciuta di tutto il genoma, su una proteina di cui si sa tutto e che rappresenta il prodotto principale della cellula che la sintetizza, e di cui è nota tutta una serie di alterazioni genetiche che presto saranno perfettamente caratterizzate a livello di sequenze nucleotidiche. Infine, l'entità degli sforzi, anche economici, dedicati alla soluzione di questo problema è ulteriormente aumentata dalla prospettiva di trovare una cura risolutiva del morbo di Cooley trasformandolo in una fenocopia HPFH. Si dovrebbe riuscire a ‛convincere' i due clusters non-α dei soggetti affetti da tale morbo, i cui geni β non funzionano, a lasciare attivi i loro geni γ, cioè a non operare il switch feto-adulto. Ed è evidente che per escogitare un sistema capace di interferire con questo switch occorre sapere come esso normalmente viene attuato.
ε) Relatività del concetto di cistrone. Si dice che due siti genetici A e B sono nello stesso cistrone quando si constata che il fenotipo di A1B1/A2B2 è diverso da quello di A1B2/A2B1, sebbene entrambi questi genotipi possiedano gli stessi alleli di A e di B, anche se assortiti in modo diverso, cioè in cis oppure in trans (donde il termine cistrone). Il significato di questo test è che essere nello stesso cistrone equivale a essere nella stessa unità funzionale. Infatti solo così si può spiegare perché, a parità di alleli, faccia differenza il modo con cui sono assortiti. Quindi il test cis-trans rappresenta uno dei modi, talora l'unico, per definire una unità di funzione genetica e per stabilire se due siti ne facciano o no parte.
Un altro criterio, usato molto comunemente, è che tutti i siti di una catena polipeptidica dipendono dallo stesso gene strutturale, cioè dalla stessa unità funzionale. Per quanto riguarda i siti genetici che controllano catene polipeptidiche diverse, non v'è dubbio che essi si trovano su geni strutturali diversi. I problemi, però, sorgono se, invece di considerare le sequenze amminoacidiche, cioè il fenotipo qualitativo, se ne prendono in esame gli aspetti regolativi, come la quantità o il momento dello sviluppo in cui sono prodotte. Infatti geni strutturali diversi, se sono molto vicini, possono appartenere a una stessa unità funzionale regolativa, cioè a uno stesso cluster o a uno stesso blocco ontogenetico. In altre parole, due siti genetici possono appartenere a due cistroni qualitativi (cioè a due geni strutturali) diversi, ma allo stesso cistrone regolativo. Questo è quanto accade ai due geni γ dello stesso cluster (che sono due geni strutturali diversi appartenenti però alla stessa unità ontogenetica, il blocco fetale) e anche - per citare un esempio celebre - ai geni dell'operone del lattosio di Jacob e Monod. Se il fenotipo viene esaminato a livello proteico qualitativo, tutti questi geni costituiscono cistroni diversi, ma se li si determina a livello ontogenetico o, rispettivamente, della inducibilità enzimatica, i geni γ da un lato, e i tre geni dell'operone del lattosio dall'altro, fanno parte di una singola unità funzionale, cioè dello stesso cistrone. In altre parole, se due siti appartengono o meno allo stesso cistrone non può essere stabilito in termini assoluti in quanto dipende dal livello fenotipico che s'è preso in esame.
Tutto ciò implica che cistroni piccoli come i geni strutturali (a loro volta costituiti da unità più piccole, le singole coppie di desossiribonucleotidi, che sono le unità di mutazione e di ricombinazione) sono contenuti in altri cistroni di ordine superiore - che ne possono contenere più di uno - che, a loro volta, sono contenuti in unità funzionali ancora più grandi, come in un gioco di scatole cinesi.
ζ) I geni ibridi. Il comportamento quantitativo dei geni ‛ibridi' dimostra che l'attività sintetica dei geni strutturali dipende da una regione alla loro sinistra e da una alla loro destra, e non solo da una regione alla loro sinistra. I geni strutturali ibridi sono stati scoperti nel 1962 da Baglioni, il quale dimostrò che l'Hb Lepore è costituita da un dimero α2 di catene α normali e da un dimero di una catena globinica lunga 146 residui amminoacidici, come la globina δ e la β, la cui sequenza è per il primo tratto identica alla catena δ e per quello restante identica alla β (v. fig. 6). La spiegazione proposta da Baglioni - dimostratasi esatta all'esame diretto del DNA - è che il gene che produce questa catena (δ-β), o Lepore, è un gene strutturale ‛ibrido' in quanto identico per il primo tratto al gene δ e per quello successivo a quello β, e che questo gene si è formato con un erossing-over ‛dislocato' (perché verificatosi tra geni non allelici, che occupano cioè posizioni diverse nel cromosoma) ma ‛omologo' (dato che questi geni sono molto simili tra loro, per cui è consentito tra di essi l'appaiamento, che è il requisito necessario affinché possa verificarsi un crossing-over tra due regioni di DNA; v. fig. 6; v. genetica: Citogenetica). In seguito sono state scoperte altre catene (δ-β) differenti per le lunghezze del tratto δ e di quello β di cui erano costituite, e anche delle catene ibride (β-δ) dette anche anti-Lepore.
Il comportamento quantitativo dei geni Lepore e antiLepore è risultato particolarmente interessante tutti questi geni, quali che siano le lunghezze dei tratti δ e β che contengono, sintetizzano all'incirca la stessa quantità di catene globiniche, e questa quantità è molto maggiore di quella prodotta da un gene δ e molto minore di quella prodotta da un gene β: se si indica con 1 l'attività sintetica di un gene δ, quella dei geni (δ-β) e (β-δ) è circa 6-7, mentre quella del gene β è circa 40. Questo dato è spiegabile solo ammettendo che l'attività sintetica dei geni δ e β dipenda da due regioni situate una alla sinistra e una alla destra di ciascuno di questi geni (oppure nella loro estremità sinistra e nella loro estremità destra). Se ci fosse una sola regione regolatrice situata alla sinistra, cioè a monte, di ciascuno di questi geni, come si verifica per i geni strutturali dei Batteri (il promotore e l'operatore sono entrambi situati a monte dei geni strutturali dello stesso operone), i geni (δ-β) dovrebbero comportarsi come i geni δ e i geni anti-Lepore come i geni β; invece, in realtà, essi si comportano come geni ‛ibridi' non solo dal punto di vista strutturale, ma anche da quello regolativo.
η) Esempi di polimorfismo bilanciato: le talassemie e la falcemia (e la enzimopenia per la G-6-PD). I dati fondamentali da cui partì Haldane nel 1949 quando avanzò l'ipotesi, ormai universalmente accettata, che la grande diffusione della talassemia fosse dovuta a una particolare resistenza dei microcitemici alla malaria perniciosa, furono: 1) la dimostrazione, fornita da Silvestroni, Bianco e Montalenti, che il morbo di Cooley era dovuto a un allele letale recessivo i cui portatori apparentemente sani sono i microcitemici; 2) la scoperta, risultato di estese ricerche popolazionistiche effettuate da Silvestroni e Bianco, che questo allele era molto frequente in alcune popolazioni italiane e virtualmente assente in altre. Haldane era stato appunto colpito da entrambe le peculiarità della diffusione della talassemia: la sua frequenza nettamente polimorfica, sebbene si trattasse di un allele letale recessivo, e la sua estrema eterogeneità interpopolazioni (che è sempre indizio di una eterogeneità altrettanto elevata nell'esposizione a un fattore selettivo).
L'‛ipotesi' di Haldane era articolata in due ipotesi parziali: a) che nelle popolazioni a elevata incidenza di talassemia gli eterozigoti per questo gene fossero avvantaggiati su entrambi gli omozigoti, cioè oltre che sugli omozigoti gravemente malati (il che era ovvio), anche sugli omozigoti per l'allele normale (e questo era tutt'altro che ovvio); b) che l'ipotetico vantaggio degli eterozigoti sui soggetti ornozigoti per l'allele normale consistesse in una loro particolare resistenza a Plasmodium falciparum.
L'ipotesi a) fu giustamente considerata da Haldane necessariamente vera; infatti frequenze di un allele letale recessivo così alte come quelle trovate in certe popolazioni per l'allele della talassemia non potevano essere spiegate altro che ammettendo che gli alleli letali recessivi, se assolutamente neutri allo stato eterozigote, si mantengano nelle popolazioni a livelli di frequenza q pari all'incirca alla radice quadrata della frequenza delle mutazioni che le producono (l'equilibrio è mantenuto quando i geni letali persi con la morte degli omozigoti, la cui frequenza è q2, sono rimpiazzati da quelli prodotti per mutazione, la cui frequenza μ è di regola circa 10-6, per cui q in genere dell'ordine di 10-3). Invece le frequenze dell'allele della talassemia arrivavano in certe popolazioni a livelli dell'ordine di 10-1, cioè circa cento volte più elevati; ciò avrebbe voluto dire che in quelle popolazioni la frequenza di mutazione, invece dei soliti valori di 10-5-10-6, era dell'ordine di 10-2 e questo dato era inaccettabile. Quindi il fattore responsabile del mantenimento, in certe aree, della frequenza dell'allele della talassemia a valori dell'ordine di 10-1 non poteva essere la mutazione; non restava allora altra possibilità che postulare l'ipotesi che in queste popolazioni gli eterozigoti per questo allele avessero una fitness (numero medio di figli fertili) maggiore di quella degli omozigoti per l'allele normale, che avrebbe bilanciato (donde il nome di ‛polimorfismi bilanciati') la sua perdita quando si trovava allo stato omozigote. Tutto questo è ormai ben noto e appare fin quasi ovvio, anche perché basato su un ragionamento matematico inoppugnabile. Ma occorreva il genio di Haldane per immaginare che un allele letale allo stato omozigote fosse addirittura vantaggioso negli eterozigoti.
L'ipotesi b), per quanto suggestiva, avrebbe invece potuto benissimo essere sbagliata. Accettata l'ipotesi a), si trattava di avanzare delle congetture sulla natura del vantaggio dell'eterozigote talassemico. Haldane si rese subito conto che il fatto che il polimorfismo per questo gene fosse limitato solo a certe popolazioni significava che lo stato eterozigote per la talassemia non conferiva ‛comunque' un vantaggio: se così fosse stato, infatti, tutte le popolazioni avrebbero presentato questo polimorfismo e non solo alcune. Haldane suppose che in alcune aree della Terra - e solo in queste - gli uomini fossero esposti a qualche fattore selettivo avverso nei confronti del quale i talassemici sarebbero stati particolarmente resistenti. Si trattava allora di individuare questo ipotetico fattore ecologico di selezione. Esso doveva avere delle connotazioni ben precise: doveva essere molto stringente e coinvolgere tutti o quasi tutti gli individui di quelle popolazioni, dato che una maggiore resistenza nei suoi confronti era stata in grado di mantenere indefinitamente e ad alta frequenza un allele letale recessivo in varie popolazioni; doveva avere avuto l'opportunità di agire per tempi lunghi su scala evolutiva; doveva aver avuto durante questi periodi una distribuzione geografica simile a quella della talassemia; doveva infine avere molto a che vedere con i globuli rossi, perché la talassemia è un fenotipo essenzialmente eritrocitario. Non v'è dubbio che questo è il ‛ritratto' della malaria perniciosa, malattia oloendemica proprio nelle aree con elevata incidenza di talassemia, che ha mietuto milioni di vittime per secoli o migliaia di anni soprattutto in età preriproduttiva (per cui una resistenza differenziale comporta automaticamente un incremento della fitness) e il cui agente causale, Plasmodium falciparum, trascorre gran parte del suo ciclo all'interno dei globuli rossi dei soggetti malati; quindi, nulla di più ragionevole che supporre che questo parassita si trovi meno bene in un globulo rosso talassemico, cioè diverso da come l'evoluzione lo ha abituato ad aspettarselo, che in un globulo rosso normale. In seguito questa ipotesi è stata estesa prima alla falcemia, cioè al gene dell'Hb S, e poi agli alleli Gd- del gene della G-6-PD.
Le prove che hanno dimostrato vera l'ipotesi di Haldane al di là di ogni ragionevole dubbio sono molte, e sarebbe troppo lungo passarle in rassegna in dettaglio; esse comprendono dati ‛microgeografici' (forte correlazione positiva tra intensità dell'endemia malarica e frequenza di questi alleli ‛malarici' anche nell'ambito di popolazioni singole relativamente omogenee per i marcatori ‛non malarici'); il riscontro dell'assenza di soggetti falcemici tra i malati con le complicazioni più gravi della malaria perniciosa, come la forma cerebrale; l'esistenza di molti alleli comuni Th e Gd-, ciascuno dei quali proprio di una popolazione che, esposta a lungo a un'intensa endemia malarica, evidentemente si è costruita per proprio conto il suo proprio o i suoi propri alleli Th e Gd- sotto la pressione di questo fattore selettivo (esempio classico di convergenza evolutiva); infine, Luzzatto ha fornito la prova diretta per l'allele GdA- sui globuli rossi di bambine negre GdA+/GdA- affette da malaria perniciosa (globuli rossi che, per il fenomeno di Mary Lyon riguardante i geni del cromosoma X come il gene Gd, costituivano una popolazione mista: alcuni avevano una normale attività G-6-PD, mentre altri erano del tutto enzimopenici), dimostrando che in ognuna di esse i globuli rossi con attività G-6-PD erano molto più parassitati dei globuli rossi enzimopenici.
In questo modo una singola spiegazione rendeva conto della variabilità intrapopolazioni (il polimorfismo era mantenuto dal vantaggio degli eterozigoti) e di quella interpopolazioni (l'assenza dell'allele talassemico nelle popolazioni non malariche dipende dal fatto che in questo caso l'eterozigote non è avvantaggiato rispetto all'omozigote normale, dato che l'essere più resistente alla malaria non è di giovamento dove non c'è malaria).
La nascita dell'ipotesi dei polimorfismi malarici rappresenta un'altra delle pietre miliari dello sviluppo della biologia, che si trovano numerose nel capitolo della genetica dell'Hb. È soprattutto per merito di quest'idea che si è inaugurato lo studio della selezione naturale a livello di singolo gene, invece che per caratteri complessi e certamente polifattoriali che non si sarebbero mai prestati a una rigorosa analisi quantitativa. La teoria sintetica della selezione naturale, in cui il darwinismo veniva finalmente integrato con la genetica, era già sorta per opera di Fisher, Wright e dello stesso Haldane circa due decenni prima (v. evoluzione), ma non si aveva un solo esempio di selezione bene analizzato a livello genetico, e non solo fenotipico, che risultasse dipendere da un singolo gene. Non solo, ma questo esempio attirava per la prima volta l'attenzione su forze selettive in un certo senso meno ‛romantiche' di quelle che erano molto di moda a quell'epoca e che si compendiavano nella famosa ‛sopravvivenza del più forte', spesso intesa un po' ingenuamente in senso letterale. Ed è noto che questo modo di travisare la selezione naturale era stato in varie occasioni strumentalizzato da varie ideologie, tanto da gettare discredito sull'intera teoria dell'evoluzione per selezione naturale. L'evoluzione biologica si svolge in genere in modo più prosaico: prevale quello che ha più figli e, per avere più figli, è molto più importante, oltre che più facile, resistere con successo a piccoli parassiti che possono causare la morte di molti individui in età preriproduttiva, che essere più forte e veloce e per questi motivi sfuggire più facilmente ai predatori. Come dice molto bene Haldane: ‟Per metterla in termini immediatamente evidenti, per un topo è molto più facile acquisire un gruppo di geni che lo renda capace di resistere a Bacillus typhimurium piuttosto di un altro che lo renda capace di resistere ai gatti".
ϑ) Le conseguenze primarie delle anomalie genetiche. Un'alterazione genetica è, per definizione, un'alterazione del DNA. Tuttavia, il livello al quale si verificano le prime conseguenze di questa alterazione, ammesso che conseguenze ci siano, può variare da caso a caso. Il sistema delle Hb offre l'intero campionario di possibilità. Ci sono casi in cui il funzionamento di uno o più geni è alterato a livello della trascrizione: per esempio nell'HPFH vengono trascritti (con tutto quel che segue) i geni γ durante la vita extrauterina: tanto l'alterazione anatomica che quella funzionale sono a livello di DNA.
È stato descritto un esempio di mutazione di un introne del βpre-mRNA, che risulta in un disturbo della sua maturazione: l'alterazione anatomo-funzionale è questa volta al livello del pre-mRNA o trascritto primario.
Poi c'è tutta una serie di anomalie genetiche in cui è alterata la traduzione, che può essere interrotta da un codone term situato in una parte iniziale della parte codificante dell'mRNA, oppure poco prima del suo termine naturale, o viceversa può non interrompersi in corrispondenza del suo punto di arresto normale. In questi casi l'espressione del gene è compromessa a livello del suo mRNA.
Esiste infine tutta la lunghissima serie delle varianti strutturali delle catene globiniche (da sostituzione di un singolo residuo amminoacidico, da delezione oppure da addizione di uno o più residui, da fusione genica, da allungamento oppure accorciamento di catena ecc.) che possono portare alle più svariate conseguenze fenotipiche, questa volta a livello proteico, a cui si è già accennato.
Nel complesso, questo sistema presenta lo spettro più completo dei modi in cui un gene può ammalarsi e dei livelli a cui può manifestare la sua malattia, e costituisce quindi un modello di grandissimo valore euristico per lo studio delle disfunzioni degli altri geni, di cui, nella quasi totalità dei casi, non si conoscono l'alterazione genetica responsabile né i meccanismi coinvolti nella disfunzione.
ι) Prospettive delle ricerche sull'Hb. Pur essendo difficile evitare in argomenti del genere una certa dose di soggettività, conviene lo stesso tentare di elencare nelle grandi linee quali potrebbero essere i prossimi sviluppi nel sistema genetico delle Hb.
A. Consensus sequences. In questi ultimi tempi si è determinata la sequenza di un gran numero di tratti di DNA appartenenti non solo ai clusters dell'Hb, ma anche a molti altri geni (v. cap. 3, È 2); la conoscenza delle sequenze non codificanti di tali geni (gli introni e le regioni fiancheggiatrici, le cosiddette flanking regions) sta portando all'identificazione delle cosiddette consensus sequences. Si tratta di sequenze che vengono individuate per due tipi di regolarità, riguardanti l'una la sequenza stessa (che si ritrova in tutte, o comunque nella grande maggioranza delle sequenze note) e l'altra la sua sede (cioè la sua distanza in termini di coppie nucleotidiche e la sua posizione a monte o a valle rispetto a un punto di riferimento ben preciso, come il segnale di inizio o di termine della traduzione o il sito di poliadenilazione, cioè il nucleotide 3′-terminale del pre-mRNA a cui viene attaccata la coda di poli-A, ecc.). La sequenza ATA, ad es., che si pensa sia il promotore dei geni strutturali, è stata individuata, a causa del suo ritrovamento costante, circa 25 coppie nucleotidiche a monte del punto di inizio della trascrizione, come anche le duplette GU e AG con cui iniziano e, rispettivamente, terminano tutte le varie decine di sequenze introniche finora esaminate. La supposizione che ognuna di queste sequenze abbia un importante significato funzionale è dovuta alle due regolarità che ne hanno permesso l'identificazione, la sequenza stessa e la sua sede, la quale ultima suggerisce inoltre la natura di questo ipotetico significato funzionale: per esempio, è naturale supporre che le duplette che delimitano gli introni siano in qualche modo coinvolte con la loro precisa rimozione dal pre-mRNA, mentre, evidentemente, è escluso che possano svolgere un ruolo nella traduzione, perché l'mRNA non le contiene. Uno degli approcci più promettenti per accertare se, e fino a che punto, le supposizioni avanzate con questi criteri siano valide è lo studio accurato, sia in vitro sia in vivo, delle conseguenze di alterazioni ben precise delle consensus sequences e/o della loro sede. Non è facile ricercare e trovare queste alterazioni, ma recentemente è stata messa a punto una tecnica che, anche se complessa e non sempre attuabile, permette di introdurre mutazioni prestabilite in punti prestabiliti di una determinata sequenza di DNA e di esaminarne poi il funzionamento a vari livelli (trascrizione, maturazione, traduzione e perfino livello proteico), cioè sulla proteina codificata dalla regione di DNA mutata in modo mirato. Quest'ultimo successo dell'‛ingegneria genetica' costituisce di sicuro uno degli approcci più affascinanti, quale nessun biofisico delle proteine di solo pochi anni fa avrebbe osato sperare di avere a disposizione, per lo studio delle relazioni tra struttura e funzione delle molecole proteiche: cosa ci può essere di meglio per affrontare questo problema che disporre di un'intera batteria di proteine alterate in punti strategici della molecola? (V. genetica: Applicazioni della genetica). Fino a poco tempo fa l'unica batteria di questo genere era costituita dalle Hb varianti e già queste erano risultate estremamente informative, ma, come è 0vvio, le mutazioni avevano prodotto queste Hb varianti a caso, non su misura per i biofisici dell'Hb.
B. I meccanismi della resistenza alla malaria. Già da qualche anno è diventato finalmente possibile mantenere in coltura il plasmodio della malaria e quindi studiare in condizioni sperimentalmente controllabili e in globuli rossi di genotipo noto e opportunamente scelto l'interazione tra questo parassita e il suo ospite, il globulo rosso. In questo modo è certo che prima o poi (le difficoltà sono ancora molte) si riuscirà a caratterizzare meglio il metabolismo intraeritrocitario dei plasmodi e a individuare cosa i parassiti si aspettano di trovare a loro disposizione nell'eritrocita, quali di queste ‛giuste aspettative' (l'evoluzione aveva loro insegnato a farvi affidamento) rimangono insoddisfatte nei globuli rossi dei soggetti resistenti e fino a che punto il plasmodio può rimediare, a livello fisiologico o evolutivo, a una ‛disillusione' di questo genere. Uno dei trucchi evolutivi messi in atto nei confronti del plasmodio sembra essere, ad esempio, un estremo accorciamento della vita del globulo rosso falcemico parassitato: il plasmodio si trova in circostanze molto sfavorevoli quando, capitato in un soggetto falcemico, deve invadere eritrociti con elevata tendenza al suicidio, che non gli concedono cioè i due giorni necessari alla sua maturazione intraeritrocitaria (la malaria perniciosa è una terzana).
Questo dei polimorfismi ‛malarici' e dei meccanismi che vi sono coinvolti è certamente il campo più avanzato di uno dei capitoli più affascinanti dell'evoluzione adattativa, l'adattamento nei riguardi di un parassita. Non si tratta semplicemente dell'adattamento a un fattore ambientale qualsiasi, per esempio climatico, ma a un fattore di selezione esso stesso suscettibile di adattarsi alle contromosse scelte dal suo ospite: una vera e propria partita a scacchi su scala evolutiva.
C. Evoluzione comparata tra regioni genetiche funzionalmente diverse. Fino a quando, e parliamo di pochi anni fa, l'evoluzione molecolare - essa stessa una conquista relativamente recente - poteva essere affrontata solo a livello di sequenze amminoacidiche, lo studio dell'evoluzione del genoma (se si esclude quella accertabile direttamente a livello cromosomico con il microscopio ottico) era soggetto a due limitazioni molto serie; infatti, potevano essere prese in considerazione solo le sequenze codificanti poiché sono le uniche che vengono tradotte in una sequenza amminoacidica, e inoltre le informazioni che si ottenevano su di esse erano incomplete (dell'evoluzione di un gene strutturale era scopribile solo quella parte che si traduceva a livello proteico qualitativo, che risultava cioè in cambiamenti amminoacidici), dato che tutti i cambiamenti sinonimi passavano, per definizione, inosservati. Per merito dell'ingegneria genetica, l'evoluzione del DNA non è più studiata per inferenza da quello che si osserva nelle proteine, ma sul DNA stesso, per cui l'evoluzione dei geni strutturali è osservabile al completo e inoltre può essere confrontata con quella di regioni di DNA di significato funzionale diverso.
Per quanto riguarda i geni strutturali si è scoperto che, sovrapposta alla loro evoluzione come regioni codificanti - il cui banco di prova è, evidentemente, la funzionalità della proteina, cioè, in ultima analisi, la sua sequenza amminoacidica - ne esiste un'altra indipendente da essa per definizione: quale tra i numerosissimi geni sinonimi (geni con sequenze diverse che, per il fatto di usare in modo diverso i codoni sinonimi disponibili, codificano catene polipeptidiche identiche) viene effettivamente adottato dall'evoluzione? Quali codoni sono prescelti per specificare un certo amminoacido tra i vari codoni sinonimi che specificano tutti quell'amminoacido? Sono state scoperte varie regolarità nella scelta di questi codoni, e di una di esse è stato possibile anche suggerire il significato evolutivo: si tratta dello scarso uso che viene fatto dei codoni che possono mutare a un codone term (risultandone l'inattivazione del gene strutturale) quando lo stesso amminoacido è codificato anche da triplette che non possono dar luogo a questo tipo di mutazioni ‛drastiche'. Anzi, si è visto anche che le mutazioni con queste conseguenze così gravi a livello di funzione genica sono rese più rare anche con un altro meccanismo, quello della preferenzialità delle sostituzioni nucleotidiche che molto difficilmente danno luogo a mutazioni drastiche.
Sempre sui geni strutturali sta venendo alla luce, proprio per i geni dell'Hb, un fenomeno del tutto imprevisto, la cosiddetta concerted evolution: si tratterebbe dell'evoluzione coordinata di due geni strettamente associati, come i due geni α o i due geni γ, grazie alla quale i due geni si sarebbero mantenuti quasi identici per tempi evolutivi durante i quali altri geni (come il δ e il β) si sono invece considerevolmente diversificati per evoluzione divergente.
Infine, è stato effettuato il confronto tra le velocità di evoluzione di regioni di DNA con significato funzionale diverso; le misurazioni sono state fatte sia in termini filogenetici, cioè tra geni omologhi di gruppi tassonomici diversi, sia per paragone tra geni non allelici ma tutti derivati da un gene ancestrale comune, come i geni γ rispetto ai geni δ e β. Tale confronto ha messo in luce che i segmenti codificanti evolvono con modalità ben diverse, soprattutto sul piano quantitativo, da quello degli introni e delle regioni esterne rispetto ai geni strutturali. Infatti, l'evoluzione di questi ultimi si svolge soprattutto per singole sostituzioni nucleotidiche (risultanti in sostituzioni codoniche sinonime e meno spesso missense, cioè causa di sostituzioni amminoacidiche) e molto meno frequentemente per delezioni e addizioni, sempre molto brevi e comunque coinvolgenti un numero intero di triplette; le altre regioni di DNA, invece, evolvono non solo per singole sostituzioni nucleotidiche, ma anche attraverso variazioni molto più estese, del tipo delle delezioni e delle duplicazioni intra- o extrageniche che possono arrivare a coinvolgere addirittura centinaia di coppie nucleotidiche. Esistono cioè modalità rapide e modalità lente di evoluzione ed è certo che il tipo di strada evolutiva che viene imboccata da una determinata regione di DNA dipende in larga misura dalla plasticità della sua struttura nei confronti della funzione che deve svolgere. L'analisi di questi differenti tipi di evoluzione probabilmente fornirà dati rilevanti per uno dei problemi più dibattuti della biologia: se l'evoluzione filogenetica delle proteine, che è quella dei loro geni strutturali, procede quasi esclusivamente a caso, vale a dire per deriva genetica, come sostengono i ‛panneutralisti', oppure sotto la guida della selezione, cioè in modo deterministico riproducibile in linea di principio, come invece sostengono i ‛panselezionisti'.
c) Genetica degli enzimi del globulo rosso.
Questi enzimi - che non sono affatto esclusivi del globulo rosso, ma in pratica vengono studiati dal punto di vista genetico solo in esso - costituiscono nel loro insieme la parte non emoglobinica del contenuto proteico del citoplasma di questa cellula. Con poche eccezioni, del tipo della catalasi e della carbonicoanidrasi, che sono molto abbondanti, ogni singolo enzima si trova nell'eritrocita in quantità così piccole - dell'ordine di poche migliaia di molecole o anche meno, in confronto ai trecento milioni di molecole di Hb - che è molto difficile studiarlo come tale, cioè come proteina. Soprattutto dal punto di vista genetico, che presuppone di regola l'esame di molti individui, si rende necessario, almeno come primo approccio, studiare non l'enzima stesso, bensì il lavoro che è capace di compiere, la sua attività enzimatica. In questo modo, infatti, si può trarre vantaggio da fattori di amplificazione molto elevati, dell'ordine del milione (ogni molecola enzimatica trasforma in 1′ qualcosa come 10-100.000 molecole di substrato, per cui in tempi dell'ordine delle ore si arriva agevolmente a cifre di questo ordine di grandezza), che permettono di studiare l'enzima in esame anche senza ‛vederlo' direttamente. È chiaro che la necessità di ricorrere a questo tipo di approccio comporta un inevitabile svantaggio: se un materiale biologico, per esempio un lisato eritrocitario, contiene un certo enzima, però in forma inattiva, esso risulta ‛assente', mentre in effetti ciò che è assente non è l'enzima bensì la sua attività. Si tratta di una situazione ben diversa da quella di proteine abbondanti come l'Hb o, nel siero, dell'albumina, dell'aptoglobina, delle transferrine ecc., che si vedono come tali, cioè indipendentemente da ‛se' e ‛come' funzionano.
È evidente che la rilevanza dell'informazione sugli enzimi che va perduta per il fatto di non poterli osservare direttamente dipende dal motivo per cui li si studia: per una ricerca genetica è molto grave non sapere se un enzima è del tutto assente o solo inattivo; viceversa questa distinzione è molto meno importante dal punto di vista clinico e farmacogenetico, e talora anche evoluzionistico, dato che almeno in molti casi queste due condizioni sono funzionalmente equivalenti.
Nella grande maggioranza dei casi due sono i caratteri che sono stati presi in esame per lo studio della genetica degli enzimi dei globuli rossi: il loro comportamento elettroforetico e la loro attività.
1. I polimorfismi elettroforetici degli enzimi del globulo rosso. - Sono questi i polimorfismi genetici che sono stati scoperti studiando su emolisati di soggetti normali un enzima ‛eritrocitario' - in realtà è ‛anche' eritrocitario - con una tecnica elettroforetica nella quale si distinguono due fasi diverse: a) migrazione elettroforetica del campione di emolisato in esame; b) evidenziazione, con una particolare tecnica di colorazione, della sede, o delle sedi, in cui si è localizzata l'attività dell'enzima in esame, che è come dire l'enzima stesso, alla fine della migrazione elettroforetica.
Prima dell'introduzione di queste tecniche di ‛amplificazione specifica', si poteva studiare il comportamento elettroforetico solo di un piccolo numero di proteine presenti in quantità molto abbondanti. Nei casi più favorevoli, come quello dell'Hb, queste proteine erano visualizzate come tali perché naturalmente colorate. In tutti gli altri casi esse erano rese visibili alla fine della migrazione elettroforetica con una generica tecnica di colorazione delle proteine in generale, con una tecnica, cioè, che né amplificava né era specifica. L'amplificazione specifica ha reso invece possibile studiare non solo un numero di gran lunga maggiore di proteine, ma anche una proteina (cioè un enzima) per volta.
Il procedimento è, almeno sul piano concettuale, addirittura elementare. Per ogni enzima per il quale si disponga di una tecnica di analisi elettroforetica adatta (condizioni di migrazione elettroforetica opportune più colorazione specifica) si esaminano gli emolisati ottenuti dai globuli rossi di un numero adeguato di individui, che costituiscono il campione in esame. Se tutti gli zimogrammi risultano uguali si conclude che l'enzima non è ‛elettroforeticamente polimorfico'; ciò significa che o esiste un solo allele del suo gene strutturale, per cui tutti gli individui della popolazione esaminata hanno lo stesso genotipo - e quindi lo stesso fenotipo elettroforetico - che è l'omozigote per questo unico allele; oppure il gene strutturale dell'enzima è sì polimorfico, ma i suoi alleli comuni sono isoelettroforetici, cioè elettroforeticamente indistinguibili. Si conclude invece che il gene strutturale dell'enzima in esame è ‛elettroforeticamente polimorfico', cioè che si è di fronte a un ‛polimorfismo enzimatico eritrocitario' (v. fig. 7), se si trovano almeno due patterns elettroforetici comuni e si dimostra che essi dipendono dal genotipo per un gene, il gene strutturale dell'enzima, in quanto uno corrisponde all'omozigosi per un allele e l'altro all'eterozigosi per questo stesso allele e per un altro allele comune anch'esso codominante; può esistere anche un terzo pattern in caso di omozigosi per quest'ultimo allele. In certi casi gli alleli elettroforeticamente distinguibili (o elettroforetici) in cui ci si imbatte comunemente sono addirittura tre, a cui corrispondono sei fenotipi o patterns elettroforetici possibili.
I risultati di questo approccio tecnico, relativamente semplice pur poggiando su basi concettuali molto solide, non si sono fatti attendere e sono apparsi addirittura strepitosi sul piano quantitativo: nel breve volgere di soli tre anni, soprattutto per opera del gruppo di Harris, esaminando 27 geni strutturali sono stati scoperti una decina di polimorfismi enzimatici eritrocitari, vale a dire una decina di nuovi polimorfismi genetici, cioè di nuovi marcatori genetici, antropologici, medico-legali, e si è così avuta la conferma di aver trovato la strada giusta per scoprirne in seguito molti altri. E tutto questo in confronto alla quindicina o poco più di polimorfismi genetici (soprattutto gruppi sanguigni) faticosamente accumulati nel corso dell'intera esistenza della genetica umana, dall'inizio di questo secolo in poi. Non solo, ma nello stesso triennio venivano con altrettanta facilità scoperti molti altri polimorfismi nella drosofila, esaminando con la stessa tecnica omogenati di questo insetto.
Ma tutto ciò rappresenta solo un aspetto secondario di questo progresso. La conoscenza di un numero molto maggiore di polimorfismi non avrebbe costituito di per sé una vera e propria svolta della genetica evoluzionistica, non solo umana, come invece è stato e per svariati motivi.
Fino a quel momento non si aveva un'idea del grado della variabilità genetica, o, peggio, se ne aveva una del tutto errata. Il motivo per cui non si poteva in linea di principio misurare la variabilità genetica, per esempio come percentuale di geni polimorfici, era che non esisteva alcun modo di individuare un gene se non attraverso la constatazione dell'esistenza di almeno due suoi alleli. Del resto, è ancora così per molti geni, come per molti gruppi sanguigni, per il gene della sensibilità alla feniltiocarbammide, per quello della visione dei colori, e così via. È evidente che, se si è capaci di scoprire un gene solo attraverso la sua variabilità, non si è in grado di determinare qual è il grado della variabilità dei geni. Ciò che è veramente importante da un punto di vista biologico è sapere quanti dei geni ‛esistenti' sono variabili. Si credeva che il grado della variabilità genetica fosse molto modesto perché, a eccezione dei gruppi sanguigni e di pochi altri caratteri polimorfici, i polimorfismi allora noti comportavano un notevole carico genetico (genetic load) in quanto uno o più dei genotipi erano francamente patologici. Basti pensare alla talassemia e alla falcemia nell'uomo e al grandissimo numero di caratteri della drosofila: per molti di questi almeno uno dei genotipi era più o meno svantaggiato. Si era quindi inevitabilmente fatto strada il concetto che ogni polimorfismo costasse, e molto, alla specie in termini di carico genetico, e che quindi nessuna specie potesse permettersi molti polimorfismi; si reputava, insomma, che quasi tutti i geni fossero monomorfici e che quei pochi che non lo erano costituissero polimorfismi bilanciati in cui fossero in gioco fattori di selezione elevati, paragonabili a quelli dei polimorfismi ‛malarici'. L'elevato carico genetico insito in tutti i polimortismi allora noti era la naturale conseguenza della rudimentalità delle tecniche allora a disposizione per cercare una variabilità fenotipica su base genetica: questa, per essere scoperta, doveva comportare quasi sempre delle variazioni accertabili a livello morfologico o clinico con conseguenze fenotipiche serie; era pertanto naturale che si fosse diffusa l'opinione che variazioni genetiche con scarse conseguenze, o addirittura prive di conseguenze, non esistessero o fossero del tutto eccezionali. In conclusione, si pensava che solo pochi dei numerosissimi geni del genoma fossero polimorfici e che quei pochissimi polimorfismi fossero soggetti a forze selettive molto intense.
Questa ben radicata convinzione è stata profondamente scossa, addirittura capovolta dai risultati dei tre anni di analisi elettroforetica delle proteine a cui si è accennato prima; adesso infatti si sa con certezza che i geni strutturali sono di regola polimorfici, e che a loro volta i polimorfismi genetici sono solo eccezionalmente soggetti a forti pressioni selettive.
I motivi di questa rivoluzione concettuale sono due: a) il fatto di avere finalmente ottenuto una stima diretta della variabilità dei geni strutturali; b) il risultato stesso della stima.
Per quanto riguarda il primo di questi due punti è evidente che per stimare il grado medio di variazione di un insieme di entità, in questo caso di geni, occorre disporre di un numero noto di geni scelti a caso e poi accertare se e quanto ciascuno di essi sia variabile. Nulla di tutto questo poteva essere effettuato solo una ventina di anni fa: ciò che si contava erano non i geni ma delle variazioni fenotipiche su base genetica, e la scelta non era affatto casuale ma effettuata sulla base della variabilità; infine su questo campione di geni, peraltro gravemente viziato e di dimensioni sconosciute, era eseguibile un'analisi poco informativa: infatti si era in grado di scoprire solo quella parte di variabilità risultante in alterazioni fenotipiche cospicue, che costituiva una quota imprecisata, anzi imprecisabile, della variabilità totale.
Tutte queste difficoltà sono state rimosse d'un colpo dall'analisi elettroforetica di un numero noto di catene polipeptidiche. Anzitutto, dato che all'inizio degli anni sessanta era ormai ben stabilita la relazione ‛un gene - una catena polipeptidica', prendere in esame n catene polipeptidiche significava prendere in esame n geni strutturali, cioè sapere quanti geni si stavano ‛studiando': era così raggiunto l'obiettivo di lavorare su un numero noto di geni scelti a caso, e anche se la scelta non era casuale in assoluto (dato che si trattava di geni amplificabili in vitro abbastanza da prestarsi a un'analisi di questo tipo), lo era - e questo è il punto di gran lunga più importante - rispetto alla eventuale variabilità.
L'analisi elettroforetica, poi, soddisfaceva perfettamente tutti i requisiti, pratici e di principio, peculiari di una tecnica utile a misurare il grado di variabilità di un numero noto di geni: i requisiti pratici, perché è molto semplice e quindi attuabile di routine su un gran numero di campioni; quelli di principio, perché la sua efficienza (probabilità che una variazione strutturale tra proteine alleliche venga scoperta, che è uguale alla proporzione tra il numero delle variazioni che verrebbero scoperte con l'elettroforesi e il numero totale di variazioni strutturali) è nota, elevata (circa 0,3) e ragionevolmente uniforme per tutte le proteine.
La variabilità dei geni strutturali è risultata essere molto grande. Dei 27 geni strutturali presi in esame dal gruppo di Harris nel triennio 1963-1966, un terzo è risultato polimorfico, pur con qualche incertezza derivante dai risultati ottenuti su popolazioni molto differenti; il grado medio di eterozigosi, praticamente identico nei Caucasici e nei Negri, era di circa 0,06, e poiché l'elettroforesi scopre circa un terzo della variabilità strutturale esistente, questi risultati implicano che i geni strutturali di regola sono polimorfici e che gli uomini sono eterozigoti per circa il venti per cento dei loro geni strutturali. Risultati molto simili sono stati ottenuti, nello stesso periodo, nella drosofila.
Anche se questi risultati riguardavano comunque solo i geni strutturali (cioè una piccola percentuale dell'intero genoma) e anzi forse solo una parte difficilmente precisabile di essi, una cosa era ormai certa: la variabilità genetica è un fenomeno molto diffuso e molto cospicuo in tutte le specie. Questa nuova nozione fondamentale rappresentava senza dubbio uno dei dati di maggior rilevanza generale emersi in un campo, come quello dello studio dell'evoluzione, fino ad allora avaro di dati di fatto di chiara e univoca interpretazione.
Sulla natura chimica di queste variazioni strutturali degli enzimi eritrocitari, anzi degli enzimi in generale, si sa tuttora molto meno, a causa delle grandi difficoltà connesse con la purificazione delle quantità che sarebbero necessarie a un'analisi delle proteine enzimatiche come tali, senza cioè poter più sfruttare la loro amplificazione funzionale come attività enzimatica. Nei pochi casi finora chiariti le differenze tra enzimi allelici sono risultate essere delle singole sostituzioni amminoacidiche che, del resto, rappresentano le variazioni strutturali di gran lunga più comuni del sistema delle Hb.
2. Variazioni su basi genetiche dell'attività degli enzimi eritrocitari. - L'attività di questi enzimi viene di solito misurata sul semplice lisato dei globuli rossi e il termine di riferimento più spesso usato è l'Hb; in certi casi, invece, l'attività viene espressa per globulo rosso. È raro scoprire con questo unico criterio una variabilità genetica unifattoriale: questo accade solo se le attività associate ad alleli diversi del gene per l'enzima in esame sono talmente diverse che classi fenotipiche discrete con attività medie diverse sono facilmente identificabili; è questo il caso della G-6-PD, di cui esistono in molte popolazioni due alleli quantitativi Gd+ e Gd- a cui sono associate attività medie di 100 (per convenzione) e, poniamo, di 10, per cui, malgrado che esista tutta una distribuzione intorno a queste medie, questi alleli sono chiaramente individuabili, specialmente nei maschi (il gene Gd è situato sul cromosoma X, per cui nei maschi esistono due sole classi genotipiche, i Gd+ e i Gd-, a cui corrispondono le attività medie di 100 e di 10, rispettivamente). In genere, invece, le attività medie associate ad alleli diversi sono sì diverse, ma con differenze molto più modeste: per esempio, all'allele più attivo è associata un'attività del cinquanta per cento superiore a quella media dell'allele meno attivo. In questi casi - che rappresentano, come si è detto, la regola - la variabilità delle singole classi fenotipiche è tale da mascherare completamente l'esistenza di classi genotipiche con attività medie diverse. È possibile allora scoprire questi alleli sulla base di altre differenze, evidenziabili individualmente perché qualitative, come le differenze elettroforetiche; quindi il confronto di gruppi - si badi bene, gruppi, non singoli individui - di soggetti tutti con lo stesso fenotipo elettroforetico, può rivelare una differenza nell'attività media, ma la sola valutazione dell'attività non permette una classificazione univoca dei singoli individui in quanto i gruppi sono distribuiti in modo ampiamente sovrapposto (v. fig. 7). La stima dell'attività media associata a un singolo allele è semplicemente la metà dell'attività media dell'omozigote per quell'allele: per esempio, se per un certo enzima A l'omozigote A1A1 mostra una attività media di 120, la stima dell'attività A associata in media a un allele A1 è 120/2, cioè 60; questa stima è di regola confermata constatando che l'attività media dell'eterozigote A1A2 corrisponde alla somma della metà delle attività medie dei due omozigoti A1A1 e A2A2. Con questo procedimento si sono confrontate le attività medie associate agli alleli comuni di un numero considerevole di enzimi eritrocitari polimorfici, e si è constatato che esse di regola sono notevolmente differenti (in media l'attività associata all'allele più attivo è di circa il cinquanta per cento superiore a quella dell'allele meno attivo: v. fig. 8). Differenze tra alleli comuni talmente grandi da far giudicare l'allele meno attivo addirittura enzimopenico rispetto a quello più attivo, pur non essendo frequenti, non sono eccezionali. Tra queste di certo le più interessanti sono le enzimopenie per la G-6-PD e quelle per alcuni enzimi responsabili della sintesi di alcuni determinanti antigenici di gruppo sanguigno, come quelle associate agli alleli i ed l (deficienze per fucosiltransferasi) e le variazioni interalleliche tra IA e IB che producono enzimi che differiscono addirittura sul piano qualitativo, cioè per il substrato che sono capaci di trasferire.
La deficienza per la G-6-PD è diffusa con frequenze molto elevate in numerose grandi popolazioni di zone con intensa endemia malarica pregressa e di essa si è avuto più volte occasione di parlare come di uno degli esempi più illustrativi di convergenza evolutiva a livello molecolare (v. anche razza).
Uno dei dati più rilevanti e istruttivi, dal punto di vista enzimologico, sulla raffinatezza dei meccanismi dell'evoluzione è emerso dallo studio delle proprietà cinetiche degli alleli enzimopenici comuni, in particolare dell'allele GdA- molto diffuso in Africa, rispetto a quelli che si trovano sporadicamente un po' dappertutto, dato che sono il prodotto diretto di mutazioni recenti. Questi ultimi sono spesso caratterizzati da proprietà cinetiche sfavorevoli che aggravano ulteriormente le conseguenze dell'enzimopenia, tanto da causare un'anemia emolitica cronica talora grave. Gli alleli Gd- comuni, quelli selezionati dalla malaria, hanno al contrario caratteristiche cinetiche tali da non causare mai una iperemolisi cronica, cioè un'anemia cronica. Le loro proprietà sono tali da assicurare un'attività G-6-PD sufficiente in condizioni normali; solo nel caso che sia richiesta a questo enzima un'attività eccezionale, per esempio per la presenza in circolo di sostanze ossidanti, esso - che già esprimeva una quota relativamente elevata della sua attività potenziale - non è in grado di aumentarla quanto sarebbe necessano; ne risulta un'insufficienza funzionale che può sfociare in una crisi emolitica in certi casi molto grave, come le ben note crisi di favismo della Sardegna, o quelle da primachina che hanno portato alla scoperta di questo tipo di enzimopenia (v. sangue: Anemie emolitiche). La lezione che si ricava da queste osservazioni è molto chiara: la selezione ha sfruttato il fatto che una normale attività G-6-PD è, in un modo o nell'altro, necessaria per uno sviluppo normale di Plasmodium falciparum, tanto che una deficienza per questo enzima può rendere più resistenti a questo agente morboso. Ma non si è accontentata di favorire alleli Gd- a caso: ha richiesto loro altre proprietà che permettessero di mantenere il vantaggio costituito dalla resistenza alla malaria senza comportare inconvenienti troppo grandi tanto a livello eritrocitario che, eventualmente, di altre cellule (il gene Gd- a differenza degli altri geni ‛malarici' noti, come il gene β, quelli delle talassemie e il Duffy, è destinato, come tutti gli altri geni house-keeping, a esprimersi in tutte le cellule dell'organismo).
Nemmeno in questo caso c'è da stupirsi dell'infallibilità dei meccanismi della selezione, che sembrano - e in effetti sono - immuni da errori grossolani come sarebbe stato quello di selezionare alleli Gd- che, pur conferendo una maggiore resistenza alla malaria, avrebbero determinato effetti pleiotropici più sfavorevoli del vantaggio conferito nei riguardi di questa malattia. La selezione non può sbagliare: sono proprio i suoi limiti che glielo impediscono. Essa guarda solo al risultato prescindendo dai modi con cui è stato conseguito, che potrebbero essere molto complicati e quindi suscettibili di ‛errate interpretazioni'. Questi sono i segreti della sua infallibilità sui grandi numeri, cioè al riparo dagli effetti del caso. Una mente ordinatrice e pianificatrice che programmi sulla base di informazioni che potrebbero essere incomplete, se non addirittura errate, può invece benissimo sbagliare, ed è questo ciò che accade nei tentativi mal riusciti di selezione artificiale.
3. La quota della variazione dell'attività degli enzimi eritrocitari che è associata alla variazione elettroforetica dei loro geni strutturali. - Su un campione di polimorfismi enzimatici eritrocitari scoperti con l'analisi elettroforetica è stata stimata la varianza dell'attività enzimatica esistente tra le varie classi fenotipiche distinguibili con l'elettroforesi; tale quota è stata poi confrontata con la varianza totale misurata direttamente su un campione di individui rappresentativo della popolazione in esame.
La varianza associata all'esistenza di un polimorfismo elettroforetico per due alleli codominanti con frequenze p e q è - a meno di un'approssimazione quasi sempre trascurabile - uguale a 2pq•d2, dove d è la differenza tra le attività associate ai due alleli espressa rispetto a quella dell'allele meno attivo (v. esempio della fig. 7). In altri termini, il contributo dato alla varianza totale dell'attività di un enzima polimorfico dalla variazione associata con questo polimorfismo è direttamente proporzionale al suo grado 2pq e al quadrato della differenza d tra le attività associate ai suoi due alleli strutturali.
Tenendo conto che circa i due terzi della variabilità dei geni strutturali, e quindi dell'attività a essi associata, sfuggono all'analisi elettroforetica, si è giunti alla conclusione che una quota considerevole, dell'ordine di un terzo, della variazione individuale dell'attività degli enzimi eritrocitari è associata alle variazioni della loro struttura.
4. Variazioni strutturali isoelettroforetiche degli enzimi eritrocitari. - Dei venti amminoacidi che costituiscono le proteine, due (l'acido aspartico e l'acido glutammico) hanno un radicale −COOH libero suscettibile di dissociarsi come anione, due (la lisina e l'arginina) hanno invece un gruppo basico, mentre tutti gli altri sono neutri; considerando la composizione media delle proteine e le probabilità delle varie sostituzioni amminoacidiche (in base al codice genetico), si arriva alla conclusione, come abbiamo già detto, che l'analisi elettroforetica dovrebbe rivelare un terzo circa delle variazioni strutturali delle proteine.
I metodi per mettere in luce le variazioni proteiche strutturali, comprese quelle isoelettroforetiche, sono diversi: per esempio, procedimenti cromatografici, di isoelectrofocusing, di valutazione della sensibilità a temperature elevate e a inibitori, di utilizzazione dei substrati, e così via. Però nessuno di questi metodi riunisce in sé tutte le prerogative dell'elettroforesi: anche quando uno di essi è stato reso talmente semplice da poter essere utilizzato in ricerche su larga scala, tanto da permettere la scoperta di nuovi alleli (isoelettroforetici) comuni, tuttavia, non conoscendone nemmeno approssimativamente l'efficienza, non è possibile valersene per verificare se la stima del grado di variazione totale dei geni strutturali raggiunta con l'elettroforesi (il triplo della variazione scoperta con questo metodo) è esatta o meno.
Questi metodi, però, hanno permesso di scoprire altri siti polimorfici all'interno di geni strutturali per i quali era già stato rivelato un sito polimorfico dall'analisi elettroforetica. In altre parole, si stanno raccogliendo esempi di geni strutturali polimorfici in più punti, e sarà presto possibile studiarne la variabilità in termini di aplotipi, cioè di assortimenti di alleli per siti variabili molto vicini invece che in termini di alleli. Dovrebbe diventare possibile accertare se e quali tipi di interazioni, a livello sia funzionale proteico sia genetico-evoluzionistico, hanno luogo tra siti polimorfici dello stesso gene strutturale di cui si conoscerà anche la sede in termini di struttura primaria proteica e nucleotidica: la delucidazione della prima deve attendere che si raggiunga anche per queste proteine lo stesso grado di conoscenze a cui si è arrivati per l'Hb; quella della seconda, che si riesca a donare e a determinare la sequenza dei loro geni strutturali. La scarsezza di queste proteine, correlata con la scarsezza dei loro mRNA, rende difficili entrambe queste linee di ricerca, ma vi sono già alcuni risultati promettenti.
L'esistenza di molti geni strutturali polimorfici in più siti fa sì che si produca nuova variabilità non solo per mutazione, ma anche per ricombinazione intragenica in doppi eterozigoti. Quanto grande sia questo contributo dipende dal grado di variabilità aplotipica (percentuale di geni polimorfici in più di un sito ed entità di questo tipo di variazione), dalle distanze medie tra siti polimorfici diversi dello stesso gene (a loro volta dipendenti anche dalla lunghezza media degli introni) e dalla frequenza media di ricombinazione per unità di lunghezza del DNA dei geni strutturali. I valori di queste grandezze sono del tutto ignoti, ma si hanno buoni motivi per ritenere che la loro risultante, cioè la variabilità prodotta per ricombinazione, sia tutt'altro che trascurabile.
5. Il problema della rilevanza biologica dei polimorfismi dei geni strutturali: la controversia tra ‛panselezionisti' e ‛panneutralisti'. - Come abbiamo già visto, è bastato constatare quanto alto fosse il grado della variabilità dei geni strutturali perché fosse definitivamente confutata l'opinione che di regola alleli diversi avessero valori selettivi molto diversi.
Ne è sorto uno dei problemi più difficili da risolvere di tutta la biologia, che può essere formulato in vari modi equivalenti: ‟Che ruolo giocano la selezione e la deriva genetica nell'evoluzione di questi polimorfismi?" o anche: ‟Quanto grande è la loro rilevanza biologica generale?" o ancora: ‟Come evolvono questi polimorfismi?"
Nessuno dubita dell'esistenza di casi in cui la selezione gioca un ruolo predominante (come nei polimorfismi ‛malarici') e di altri, come nella maggioranza, se non in tutti, i polimorfismi ‛privati' (cioè limitati a un piccolo gruppo isolato), in cui è invece la deriva genetica il fattore evolutivo principale. Il difficile, però, è di accertare il comportamento ‛generale' dei polimorfismi, non quello di alcuni casi modello. Due semplici considerazioni rendono evidente come le difficoltà di accertarlo siano quasi insormontabili.
α) Anzitutto, è certo che di regola i fattori selettivi in gioco in ogni singolo polimorfismo sono molto piccoli, se non addirittura nulli. Secondo i ‛panselezionisti' la selezione gioca comunque un ruolo in tutti i polimorfismi, sia pure con variazioni minime della fitness. I ‛panneutralisti' sostengono invece che gli alleli comuni dei geni polimorfici sono del tutto neutrali. Sia per gli uni sia per gli altri la selezione svolge un compito negativo fondamentale funzionando da filtro contro gli alleli incondizionatamente sfavorevoli (che sono verosimilmente la maggioranza) prodotti di continuo per mutazione e ricombinazione intragenica. È invece sul suo ruolo nei riguardi degli alleli comuni che le due vedute divergono in modo sostanziale: per la prima, quella dei ‛panselezionisti', questi alleli sono diventati comuni perché conferivano un vantaggio, cioè per opera della selezione; per la seconda, quella dei ‛panneutralisti', solo per effetto del caso. Non bisogna lasciarsi ingannare dalle apparenze e dire che in fondo queste due visioni del modo di procedere dell'evoluzione differiscono di poco, perché perfino i ‛panselezionisti' sono convinti che le differenze di fitness tra i vari genotipi devono essere comunque molto piccole, e concludere che tra ‛molto piccole' e ‛zero' non ci corre molto. Infatti i geni polimorfici sono numerosissimi, per cui un effetto piccolo per gene può produrre grossi risultati sull'insieme. Ma, a parte questa considerazione, ciò che rende queste due concezioni fondamentalmente diverse, si direbbe a un livello perfino filosofico, è che per la teoria panselezionista l'evoluzione dei geni strutturali è un fenomeno deterministico, cioè in linea di principio e su grandi numeri predicibile; secondo la teoria panneutralista, invece, si tratterebbe di un fenomeno essenzialmente stocastico, dato che procederebbe ‛fissando a caso alleli neutrali prodotti a caso'. Per la teoria panselezionista i polimorfismi esistenti sarebbero o polimorfismi bilanciati o polimorfismi transeunti che viaggiano verso la fissazione dell'allele più favorevole con un andamento in linea di principio riproducibile; per quella panneutralista tutti i polimorfismi sarebbero transeunti e in uno ogni tanto, a caso, si fisserebbe, sempre a caso, uno dei suoi alleli comuni.
β) L'efficienza della misura diretta della fitness è bassissima. Per definizione, i fattori selettivi o coefficienti di selezione sono differenze tra i valori delle idoneità genotipiche; essi devono perciò essere valutati in modo attendibile. Ebbene, la dimostrazione diretta che l'idoneità degli eterozigoti per la falcemia è maggiore di quella degli omozigoti per l'allele βA (tanto per citare l'esempio più noto) non è stata mai data, sebbene si sia certi che questa differenza esista e sia molto grande, cioè circa 0,1 (cioè l'idoneità degli omozigoti normali è di circa il dieci per cento più bassa di quella dei soggetti falcemici in zone fortemente malariche in cui l'allele βS ha una frequenza stabile di circa 0,1). Questo non deve stupire: basta pensare alle grandi difficoltà pratiche che si incontrerebbero se si volesse dimostrare in modo rigoroso che effettivamente in tali zone i soggetti con la sola Hb A hanno in media un numero di figli che è il 90% del numero medio di figli dei soggetti con l'Hb A e l'Hb S. Con questo non si vuole affatto sostenere che non si è raggiunta la certezza che lo stato eterozigote per la falcemia protegga dalla malaria perniciosa; abbiamo solo voluto dare una misura dell'estrema insensibilità dei metodi di misurazione diretta della fitness, che non sono riusciti a dimostrare l'esistenza di un fattore selettivo nemmeno nel caso in cui era all'opera una selezione considerevolmente intensa, e lo si sapeva a priori con certezza.
In conclusione, quindi, il problema del ruolo della selezione nell'evoluzione dei geni strutturali nel loro insieme non può essere risolto in modo diretto; non è cioè possibile confrontare le idoneità dei genotipi comuni per molti geni polimorfici, per accertare per quanti di essi le idoneità dei loro genotipi sono diverse (geni soggetti anche alla selezione oltre che alla deriva genetica) e per quanti le idoneità di tutti i genotipi comuni sono identiche (geni polimorfici che evolvono solo per deriva genetica). Chiaramente, non è possibile cercare differenze di idoneità sapendo che, se esistono, hanno valori tutt'al più dell'ordine dell'1%, e usando un metodo che non è in grado di scoprire una differenza del 10% nemmeno sapendo in anticipo che tale differenza esiste. Per la determinazione della percentuale dei geni polimorfici si dispone di un metodo sensibile ed efficiente; ma per determinare quale percentuale di geni polimorfici sia soggetta anche alla selezione non si dispone di alcun metodo diretto efficace. È quindi necessario ripiegare su metodi indiretti.
Se due alleli di un gene strutturale hanno valore selettivo (cioè non sono equivalenti o neutrali) è segno che producono proteine alleliche funzionalmente tanto diverse e/o in quantità così differenti da avere effetti diversi sull'idoneità degli individui con genotipo differente per questi alleli. L'evoluzione di questo gene ne verrà allora influenzata e in media sarà diversa da quella dei geni che evolvono solo per deriva genetica. Gli approcci indiretti per chiarire se un polimorfismo sia soggetto anche alla selezione possono essere quindi raggruppati in approcci a monte e a valle dell'eventuale effetto sulla fitness.
Fanno parte del primo gruppo gli studi volti ad accertare se e in quanti enzimi polimorfici, in un campione di polimorfismi scelti a caso, le variazioni strutturali scoperte con l'elettroforesi sono associate a variazioni di proprietà cinetiche funzionalmente significative che sia ragionevole supporre abbiano un certo effetto sul modo di funzionare delle cellule dove risiedono questi enzimi. Questi dati sono solo indicativi, perché non è affatto detto che una variazione, poniamo dell'attività enzimatica, abbia conseguenze tanto cospicue da influenzare in modo evolutivamente significativo un carattere complesso come l'idoneità riproduttiva dell'intero organismo. Potrebbe essere infatti che la variazione interallelica di attività osservata in vitro non si verifichi in vivo, cioè in condizioni biochimiche ben diverse; oppure che questa variazione si verifichi sì anche in vivo, ma che sia irrilevante sul piano fisiologico; oppure ancora che ci sia una variazione a livello fisiologico complesso, ma che sia perfettamente compensata dall'organismo, per cui essa risulti di fatto irrilevante per la sua idoneità. Esistono tuttavia due risultati interessanti che, nonostante questa serie di incertezze, suggeriscono che il modo di evolvere dei polimorfismi enzimatici ‛eritrocitari' sia in una certa misura guidato dalla selezione: 1) ad alleli comuni diversi dal punto di vista elettroforetico (cioè strutturale) sono in genere associate attività enzimatiche considerevolmente diverse (v. fig. 8); 2) in quasi tutti i polimorfismi enzimatici in cui le attività associate ai vari alleli comuni sono diverse, l'allele con attività più elevata è il più frequente.
Gli approcci a valle si fondano sul fatto che l'evoluzione è un processo additivo, nel senso che gli effetti dell'eventuale esistenza di diversità di fitness tra genotipi differenti si sommano di generazione in generazione. Quindi differenze di idoneità di per sé non constatabili possono essere amplificate nel corso delle generazioni fino a dar luogo a effetti facilmente apprezzabili. In assenza di selezione, i polimorfismi genetici evolvono solo per effetto della deriva genetica ed eventualmente della genetic admixture o commistione genetica (v razza), cioè di due fattori che, a differenza della selezione, agiscono su tutti i geni con la stessa intensità. Quindi se, misurando per tutta una serie di polimorfismi l'effetto di questi fattori (quello della deriva genetica è espresso come ‛distanze genetiche' e quello della genetic admixture come grado di commistione, m), si constata che per alcuni di essi si trovano valori diversi da quelli relativamente uniformi di tutti gli altri, si deve concludere che la loro evoluzione è stata influenzata in larga misura anche dalla selezione. E questo è appunto quanto si osserva con i polimorfismi malarici, per i quali si trovano distanze genetiche molto più grandi di quelle che si osservano per gli altri polimorfismi e indici (apparenti) di commistione genetica m del tutto anormali.
Tanto gli approcci diretti che quelli indiretti hanno prodotto risultati molto interessanti, ma certo non risolutivi per la questione del modo di evolvere dei geni strutturali, cioè delle proteine, e verosimilmente ci vorrà molto per risolvere questo problema di fondo della biologia. Del resto gli evoluzionisti, per la natura stessa del problema (storico) di cui si occupano, non sono abituati ai ritmi trionfali della biologia molecolare: i tempi con cui vengono scandite le principali tappe dei loro progressi (pubblicazione dell'Origine delle specie, 1859; teoria sintetica dell'evoluzione, anni trenta; ipotesi dei polimorfismi ‛malarici', 1949; scoperta che la variabilità genetica è molto grande, 1963-1966, cioè una pietra miliare ogni venticinque anni in media) sono ben più lenti. Tuttavia un altro grande successo sta delineandosi: si tratta della determinazione del grado e delle carattehstiche (proprietà descrittive) della variabilità del DNA non codificante, cioè per la prima volta di regioni di genoma al di fuori dei geni strutturali e comunque non studiate attraverso l'intermediarietà delle proteine.
d) Genetica dei gruppi sanguigni.
Ogni carattere del sangue che presenti un polimorfismo genetico, che dipenda cioè da due o più alleli comuni che permettano di suddividere individui della stessa specie in almeno due ‛gruppi', può essere considerato in linea di principio un carattere di gruppo sanguigno. Poiché però per quasi mezzo secolo gli unici polimorfismi genetici del sangue noti erano quelli degli antigeni eritrocitari, è ormai invalso l'uso di riservare questo termine solo a essi.
Per ‛sistema' di gruppo sanguigno si intende l'insieme di tutti gli antigeni di gruppo che dipendono dallo stesso gene o dallo stesso blocco di geni. Tutti i sistemi e antigeni di gruppo sanguigno sono stati quindi scoperti con tecniche sierologiche, per definizione. Per alcuni di essi si è intrapreso in seguito anche uno studio biochimico che ha reso conto in termini molecolari, integrandole, di molte delle relazioni formali scoperte a livello sierologico. La maggior parte di tali sistemi si trova ancora in questa prima fase, perché la loro natura chimica ci è del tutto ignota. Sarebbe quindi un grave errore, sopravvalutando i successi ottenuti grazie allo studio biochimico di alcuni antigeni di gruppo sanguigno, considerare ormai superata la loro descrizione formale a livello sierologico; infatti essa sola può fornire le indispensabili indicazioni all'approccio biochimico, come è già avvenuto per i pochi sistemi chiariti a livello molecolare.
1. Livello sierologico. - A. Tecniche di laboratorio: l'agglutinazione. La tecnica fondamentale utilizzata per lo studio di tutti gli antigeni di gruppo sanguigno consiste nell'osservare se, mescolando con un antisiero una sospensione di globuli rossi, questi si agglutinano. Quando questo accade, è spesso utile determinare il titolo dell'antisiero, poiché in molti casi i sieri presentano un titolo tanto più elevato quanto più ricchi dell'antigene corrispondente sono i globuli rossi con cui li si titola, e diventa quindi possibile in certe condizioni misurare in modo approssimato le quantità - si noti bene: relative - di un dato antigene presenti in eritrociti di soggetti diversi.
Alcuni di questi antigeni possono esistere in soluzione nella saliva e in altri liquidi dell'organismo, dove possono essere ricercati sfruttando la loro capacità di inibire la reazione di agglutinazione corrispondente: per esempio, una saliva con antigene A è in grado di inibire, spesso anche se molto diluita, l'agglutinazione di globuli rossi A da parte di un siero α (anti-A).
B. Procedimenti concettuali. L'interpretazione dei risultati relativi ai gruppi sanguigni deve seguire un suo iter particolare molto più complicato di quello diretto che si può adottare per i polimorfismi biochimici. Infatti esiste in questo caso un'unica tecnica di base, l'agglutinazione, che è uguale per tutti i sistemi. Anche nel caso dell'analisi elettroforetica, una parte del procedimento, la migrazione o corsa elettroforetica, è comune a tutti gli enzimi, ma alla fine di questa si usa un reagente che mette in evidenza il solo enzima in esame, per cui qualsiasi variazione genetica si osservi nello zimogramma è da attribuirsi senz'altro a quell'enzima. Per i gruppi sanguigni, mancando una tecnica propria di ogni sistema, l'unico strumento di cui si dispone per stabilire se un determinato antigene corrisponde a uno di quelli già noti o - nel caso risulti essere un nuovo antigene - se fa parte di un sistema conosciuto o se è invece il primo antigene di un nuovo sistema, è l'analisi formale, cioè statistica, dei dati, che può essere condotta tanto a livello popolazionistico che a livello famigliare.
α) Come sono state scoperte le molte decine di antigeni eritrocitari che si conoscono attualmente. La scoperta del sistema AB0 da parte di Landsteiner all'inizio di questo secolo fa storia a sé non solo perché ha dato origine a questo capitolo della biologia, rendendo possibile per la prima voIta suddividere gli uomini in gruppi sanguigni diversi, i ben noti 0, A, B e AB (v. immunologia e immunopatologia), ma anche perché esistono in questo sistema peculiari agglutinine, dette ‛naturali' perché non indotte da trasfusioni di sangue antigenicamente diverso. Poiché il siero di ciascun individuo contiene tutte le agglutinine (anticorpi) corrispondenti agli agglutinogeni (antigeni) del sistema AB0, che mancano nei suoi globuli rossi, è stato sufficiente cimentare i sieri di un certo numero n di individui presi a caso, uno a uno, con i globuli rossi di ciascuno di questi stessi individui (per un totale quindi di n2 prove) per scoprire questo sistema e rendersi conto che esso consisteva di due agglutinogeni (detti A e B) e di due agglutinine (dette α e β).
Nessuno di tutti gli altri sistemi di gruppo sanguigno è stato scoperto in questo modo. Né avrebbe potuto esserlo: infatti, se si cimenta il siero di un individuo con i globuli rossi di un altro appartenente allo stesso gruppo secondo il sistema AB0, non si verifica agglutinazione, anche se i due soggetti hanno gruppo sanguigno diverso per uno o più degli altri sistemi. Infatti, di regola non si possiedono, come per l'AB0, agglutinine dirette verso gli agglutinogeni assenti nei propri globuli rossi: è questo, naturalmente, il motivo per cui, riguardo alla trasfusione, il sistema AB0 è molto più importante di tutti gli altri sistemi messi insieme.
Tralasciamo per brevità la storia delle scoperte dei sistemi MN e P (a cui si è arrivati immunizzando conigli con globuli rossi umani con lo scopo deliberato di trovare antigeni di gruppo indipendenti dagli unici allora noti, quelli del sistema AB0) e consideriamo tutti gli altri antigeni e sistemi di gruppo sanguigno. Ebbene, salvo differenze che dal punto di vista teorico sono di dettaglio, la grande maggioranza di queste scoperte ha seguito il seguente iter obbligato, a inizio sempre fortuito: a) il siero di un soggetto che deve ricevere una trasfusione di sangue (quasi sempre un politrasfuso) risulta, contrariamente alle previsioni, capace di agglutinare i globuli rossi di un altro che, compatibile con esso per il sistema AB0, si presumeva fosse un donatore adatto. A parte il problema pratico di trovare un altro sangue adatto alla trasfusione, si cerca di chiarire, anche da un punto di vista teorico, la natura dell'agglutinina o delle agglutinine la cui presenza nel siero del ricevitore era inattesa; b) anzitutto si determina il fenotipo sierologico (cioè per tutti gli antigeni eritrocitari noti) dei globuli rossi dei due soggetti: l'antigene riconosciuto dall'agglutinina inattesa deve mancare nel sangue del ricevitore ed essere presente in quello del donatore (naturalmente, se quest'ultimo è in realtà composto da un miscuglio di sangui di diversi tipi il problema diventa più complesso ma resta tuttavia risolvibile). Talora già a questo stadio si arriva molto vicino alla soluzione: questo accade se uno dei due soggetti presenta un fenotipo eccezionale per un sistema, che è quindi presumibilmente quello coinvolto nell'inattesa reazione di incompatibilità; c) si cimenta quindi il siero del ricevitore con globuli rossi che presentano opportune costellazioni degli antigeni non esclusi nella fase b) e ci si accerta se esso contenga una agglutinina diretta verso uno di questi antigeni noti, ma di cui normalmente non si tiene conto nelle trasfusioni. Se, come in genere accade, si identifica questa agglutinina, il problema è risolto e si è trovato un altro esempio di agglutinina rara (per esempio un anti-e). Se invece la capacità agglutinante inattesa del siero in esame non è attribuibile a nessuna specificità nota, si deve concludere di aver scoperto una nuova agglutinina e con essa il corrispondente agglutinogeno eritrocitario.
Alcuni particolari alleli molto rari, ma di grande interesse teorico, sono stati invece scoperti attraverso uno studio approfondito di casi di apparente incompatibilità tra un genitore (talvolta la madre) e un figlio. Tra i più interessanti: il ‛cromosoma' Mk che non produce né l'antigene M, né l'N, né l'S, né l's; e soprattutto il gene IAB che conferisce la capacità di sintetizzare l'antigene A e l'antigene B.
Anche ricerche antropologiche hanno talora portato alla scoperta di fenotipi eccezionali, come il fenotipo Rhnull (assenza completa di tutti gli antigeni del sistema Rh, inferita dal fatto che questi globuli rossi non risultano agglutinabili da alcuno degli antisieri del sistema Rh finora noti), di estremo interesse per la comprensione del sistema Rh.
β) Procedimento generale nello studio della genetica di ciascun antigene eritrocitario considerato isolatamente. Di regola si comincia con la stima della sua frequenza in una o più popolazioni. Se cimentando il siero del ricevitore con i globuli rossi di un adeguato numero di soggetti (qualche centinaio) si trovano tra di essi tanto dei positivi che dei negativi, si può concludere che l'antigene in esame è un antigene polimorfico, cioè un antigene di gruppo. Talora si trova invece un unico tipo di risultato: tutti positivi (in questo caso quell'antigene, che tutti possiedono salvo il ricevitore, rientra nella classe dei cosiddetti antigeni ‛pubblici') o tutti negativi (e allora quell'antigene di cui quasi tutti sono privi - con l'eccezione, naturalmente, del donatore - rientra nella classe dei cosiddetti antigeni ‛privati').
La prima informazione sulla genetica di questo antigene la si ottiene direttamente dall'esame di un campione di soggetti scelti a caso: dato che di regola la presenza di un antigene è sotto controllo di un gene, se le frequenze relative dei soggetti positivi e di quelli negativi sono le stesse nei due sessi, è praticamente certo che l'antigene dipende da un gene autosomico. Se invece queste frequenze sono diverse e in particolare se quella delle femmine negative è più bassa di quella dei maschi negativi (più precisamente, è uguale al quadrato di quest'ultima), allora è molto verosimile che l'antigene dipenda da un gene del cromosoma X (questo si è verificato finora solo per il sistema Xg).
Ma, a parte questa informazione, la genetica di un antigene si può studiare solo esaminando un adeguato numero di famiglie. Tutte le volte che questo è stato fatto è risultato che l'antigene dipendeva da un allele capace da solo di produrre quantità sufficienti a renderlo evidenziabile con la normale tecnica dell'agglutinazione (incroci tra individui positivi danno origine a figli positivi e a figli negativi, mentre se tutti e due i genitori sono negativi tutti i figli sono negativi). Spesso si può dimostrare anche il cosiddetto ‛effetto dose (genica)': i globuli rossi di soggetti certamente eterozigoti mostrano di contenere meno antigene rispetto ai globuli rossi dei soggetti omozigoti.
A questo punto è praticamente conclusa la prima fase dello studio genetico di questo antigene e può cominciare quella, molto più complessa, di accertare le sue eventuali relazioni con sistemi già noti.
γ) Procedimento generale per accertare se esistono relazioni tra un antigene e gli altri, e la loro natura. L'unico approccio utilizzabile consiste nell'accertare per ‛ogni antigene noto' se, a livello popolazionistico, mostra una distnbuzione indipendente da quella dell'antigene in esame e se, a livello famigliare, segrega in modo indipendente da esso o per lo meno ricombina con frequenza apprezzabile. Supponiamo ad esempio di chiamare X l'antigene in esame e che si vogliano esplorare le sue eventuali relazioni con gli antigeni, 1, 2, 3 ... n, già noti. Ebbene, per studiare X rispetto a 1 occorrerà esaminare gli stessi soggetti con un siero anti-X, che permetterà di classificarli in due gruppi: X(+) e X(−), e con un siero anti-1, suddividendoli quindi in 1(+) e 1(−) (v. tab. IV). Lo stesso vale naturalmente per gli studi famigliari. Si tratta in sostanza di stabilire se la frequenza degli 1(+) trovata all'interno della classe X(+) si possa ragionevolmente considerare uguale e quella degli 1(+) trovata nella classe degli X(−). Lo strumento statistico che si utilizza a tale scopo è la tabella di contingenza (che, senza dubbio, per la costruzione dell'intero capitolo dei gruppi sanguigni ha giuocato un ruolo assolutamente preminente) e la significatività degli eventuali scarti tra frequenze attese e osservate si determina con il χ2.
Gli studi famigliari si effettuano con i metodi della genetica classica, per l'analisi di pedigree in cui segregano contemporaneamente due o più caratteri di ciascuno dei quali, preso singolarmente, si deve conoscere la genetica formale.

C. Risultati: la genetica formale dei gruppi sanguigni e le sue implicazioni. Sono state finora scoperte decine di antigeni di gruppo sanguigno, più un gran numero di antigeni pubblici e di antigeni privati, per molti dei quali non è ancora chiaro a quale sistema appartengano o se siano i primi rappresentanti di un nuovo sistema.
Dallo studio degli antigeni di gruppo, cioè polimorfici, che via via si andavano scoprendo, nelle loro relazioni con gli antigeni già noti, sono scaturiti risultati che nel loro complesso hanno portato al quadro attuale di quindici sistemi chiaramente individuati, alcuni dei quali molto complessi, come l'Rh, e altri che, pur essendo indipendenti dal punto di vista genetico, interagiscono fra loro in modi che sono stati poi confermati e chiariti a livello biochimico.
Un antigene X (e l'allele che lo produce) può, rispetto a tutti gli altri già noti, mostrare le varie caratteristiche di seguito illustrate.
α) Essere indipendente tanto a livello popolazionistico che famigliare: si conclude, in questo caso, di aver scoperto un nuovo sistema di cui X è per il momento l'unico rappresentante. Il sistema consiste, in questa fase, di un gene con due alleli, uno dei quali produce anche in singola dose l'antigene X e l'altro non lo produce; l'unico mezzo per studiarlo è il siero anti-X. Due soli sono quindi i fenotipi: X(+) e X(−), e tre i genotipi, l'omozigote e l'eterozigote per l'allele che produce X (fenotipo X[+]) e l'9mozigote per l'allele che non produce X (fenotipo X[−]). È evidente che, definendo in questo modo (che è l'unico possibile) i fenotipi di questo nuovo sistema X, l'allele che non produce l'antigene X è necessariamente considerato ‛recessivo', mentre quello che lo produce appare ‛dominante'. Però di regola si verifica che l'allele che non produce X produca un altro antigene allelico a X, cioè che questo allele non sia affatto silente e quindi intrinsecamente recessivo, bensl che produca un antigene che per caso non è stato scoperto. Se quest'ultimo antigene fosse stato il primo a essere scoperto, sarebbe questo allele ad apparire dominante e l'altro ancora sconosciuto ad apparire recessivo. Inoltre l'allele che non produce X potrebbe essere in realtà un insieme di alleli a ciascuno dei quali corrisponderebbe un antigene allelico a X. A livello di nomenclatura tutto ciò ha avuto la sua conseguenza: è questo, ad esempio, l'unico motivo per cui all'antigene C, scoperto per primo, è stata riservata la lettera maiuscola, e all'antigene allelico prodotto da un allele anch'esso codominante come C, ma scoperto solo in seguito, è stato assegnato il simbolo c.
A eccezione dei sistemi AB0 ed MN, lo studio di tutti i sistemi attualmente noti è passato per questa fase iniziale. Alcuni ci si trovano tuttora. Il più noto tra questi è il sistema Xg con due alleli Xga e Xg e quindi due fenotipi: Xg(a+), a cui corrispondono nei maschi il genotipo Xga e nelle femmine i genotipi XgaXga e XgaXg, e Xg(a−), a cui corrispondono nei maschi il genotipo Xg e nelle femmine il genotipo XgXg. Non è noto se Xg, che certamente non produce l'antigene Xga, produce un antigene o se è un allele silente, cioè intrinsecamente, e non solo apparentemente, recessivo.
β) L'antigene X può essere l'‛unico' antigene allelico di un antigene già noto. In questo caso, molto comune e di facile soluzione, non si trovano soggetti che manchino di tutti e due questi antigeni, pur essendo frequenti quelli privi di uno dei due (gli omozigoti per uno dei due alleli).
La scoperta dell'anti-c può illustrare questa situazione (ma si potrebbero fare molti altri esempi): quando si è scoperto l'antigene che è poi stato chiamato c, si è capito che esso era l'unico antigene allelico (comune) di C perché non si sono trovati individui negativi sia con questo nuovo siero (ancora anti-X) sia con l'anti-C, mentre, se i due antigeni fossero stati controllati da due geni diversi, circa il 6% degli individui in esame avrebbero dovuto essere C(−) e X(−) (i[C−] erano circa il 32,5% e gli X[−] circa il 18,5%). La conferma definitiva si è poi avuta constatando che le frequenze delle tre classi fenotipiche osservate (C[+]c[−]=CC), (C[+]c[+]=Cc) e (C[−]c[+]=cc) corrispondevano a quelle attese dalla legge di Hardy-Weinberg (v. evoluzione) per un locus con due alleli codominanti. Inoltre, i c(+) che erano anche C(+) (che cioè possedevano un solo allele c) mostravano di avere meno antigene c rispetto ai soggetti C(−) che sono omozigoti per c. Vale la pena di notare ancora una volta che un risultato così netto, conclusivo e semplice da ottenersi e da interpretarsi era raggiungibile solo a patto di usare i due sieri sugli ‛stessi' soggetti. Ciò illustra una peculiare difficoltà della genetica formale dei gruppi sanguigni rispetto a quella basata sull'analisi elettroforetica degli enzimi già illustrata: la ricerca di antigeni allelici richiede reagenti (cioè antisieri) specifici diversi, esattamente come la ricerca di antigeni controllati da geni indipendenti; con una prova si cerca un antigene e con un'altra, cioè con un altro antisiero, l'altro antigene. È chiaro che se il soggetto in esame è portatore di un antigene allelico diverso dai due cercati con queste due prove esso non viene scoperto, e solo l'uso di un ulteriore antisiero lo può evidenziare. Ed è chiaro altresì che non vi è a priori nessun indizio per sospettare quale sia fra gli antisieri noti quello che reagisce con un antigene allelico all'antigene che si sta studiando. Viceversa, ogni singola analisi elettroforetica specifica per un enzima è in grado di individuare ‛tutti' gli alleli elettroforetici di quell'enzima.
γ) L'antigene X può essere allelico a un antigene già noto. In questo caso i risultati più informativi sono quelli famigliari, che mostrano facilmente il rapporto di allelismo. Per esempio, si è arrivati alla conclusione che gli antigeni A e B dipendono da due alleli quando ci si è accorti che coloro che li possiedono entrambi ne trasmettono sempre uno a ciascuno dei loro figli, cioè in nessun caso tutti e due o nessuno dei due.
δ) L'antigene X può essere un antigene di sottogruppo. Il siero chiamato anti-A1, ad esempio, rende possibile la suddivisione in due gruppi, i positivi e i negativi, ma solo nell'ambito degli individui che hanno l'antigene A, mentre quelli che non lo possiedono (i B e gli 0) sono tutti negativi. In altri termini, l'antigene individuato da questo siero può essere presente solo in soggetti che hanno l'antigene A. Il gene IA1 responsabile della comparsa di questo antigene, oltre che dell'antigene A, è uno degli alleli del sistema AB0; è dominante sull'allele i e sull'IA2 che produce soltanto l'antigene A e codominante rispetto all'allele IB.
ε) L'antigene X può far parte di un sistema pur senza essere allelico degli antigeni di quel sistema. Questo particolare tipo di relazione tra marcatori genetici fu intuito per la prima volta all'inizio degli anni quaranta da Fisher e Race, che coniarono il termine di ‛cromosoma Rh' per indicare una regione genetica che consisteva di tre marcatori (cioè di tre siti di ciascuno dei quali erano stati individuati due alleli) associati in modo talmente stretto da non mostrare mai ricombinazione a livello famigliare e presentare a livello popolazionistico un fortissimo linkage disequilibrium, cioè una distribuzione degli 8 diversi possibili assortimenti (2 per ciascuno dei tre marcatori=23) o ‛cr0mosomi' o, come si preferisce chiamarli ora, aplotipi, molto lontana da quella casuale. Pochi concetti sono stati piu fecondi di questo per la genetica umana, anzi per la genetica in generale. Al ‛cromosoma Rh' sono seguiti l'MNSs, il Kell, il Gm, l'HLA e il blocco dei geni non-α, alcuni dei quali costituiscono tuttora i modelli più illustrativi di cui si disponga, negli Eucarioti, di clusters di geni le cui funzioni sono strettamente correlate e coordinate.
Non è il caso di esaminare in dettaglio i risultati che sostengono questa ipotesi ormai universalmente accettata, almeno nelle sue linee generali. Vale però la pena di considerare i più dimostrativi. Esistono tre marcatori ben individuati, ciascuno con due alleli comuni; il primo marcatore è D e d: D produce l'antigene D o Rh anche in singola dose, per cui i soggetti Rh(+) sono genotipicamente DD o Dd, mentre i dd sono Rh(−). Gli altri due marcatori, C e c, E ed e, sono allelici due a due e tutti codominanti e le distribuzioni dei tre fenotipi possibili per Cc e per Ee sono in accordo con la legge di Hardy-Weinberg. Tuttavia, si è osservato che esistono deviazioni fortissime (la più piccola di un fattore 6 - che non è poco - e la più grande di circa 350 volte) di tutte le distribuzioni fenotipiche di questi tre marcatori (per es. tra gli Inglesi, i CC, che tra gli Rh[+] sono circa il 20%, tra gli Rh[−] sono solo 1 su circa 1.700 persone) considerati due a due, rispetto a quelle che si avrebbero se la frequenza di ognuno di questi antigeni fosse indipendente da quella degli antigeni degli altri due marcatori. Fisher e Race hanno spiegato questi dati postulando l'esistenza di 23, cioè 8, ‛cromosomi', ciascuno dei quali con uno dei due alleli di ognuno dei tre marcatori e con frequenze ben lontane dall'equilibrio, cioè dal prodotto delle frequenze degli alleli in esso contenuti. Ognuno di questi ‛cromosomi' è ereditato ‛in blocco', cioè con una frequenza di ricombinazione al suo interno così bassa da apparire uguale a zero in studi famigliari e da prevenire il raggiungimento di frequenze casuali dei vari assortimenti allelici o ‛cromosomi' (o aplotipi) a livello popolazionistico (v. fig. 9).
Era così sorto il concetto di linkage disequilibrium, uno dei più fondamentali della genetica: gli alleli di siti strettamente linked sono di regola assortiti non a caso, perché il tempo necessario a raggiungere l'assortimento casuale partendo - come necessariamente accade - da assortimenti non casuali è lunghissimo. Infatti questo risultato finale richiede che essi ricombinino (nei doppi eterozigoti) e, tra siti strettamente associati, questo evento è, per definizione, molto raro.
Ciascuno di questi 8 ‛cromosomi Rh' si comporta come un allele a livello famigliare e anche a livello popolazionistico (se si considerano tempi brevi in termini evolutivi), per cui i loro vari assortimenti genotipici, cioè a due a due, si presentano di regola con le frequenze previste dalla legge di Hardy-Weinberg. Ad esempio, il genotipo DCe/DcE ha in Inghilterra la frequenza di 0,11, che è pari al doppio prodotto delle frequenze di questi due ‛cromosomi' in questa regione (2×0,4×0,14=0,112). È così possibile stimare le frequenze di questi 8 ‛cromosomi' partendo dalle frequenze dei vari fenotipi Rh: per esempio, la stima della frequenza del ‛cromosoma' dce è la radice quadrata della frequenza dei soggetti D(−)C(−)c(+)E(−)e(+), i soggetti, cioè, certamente omozigoti per questo ‛cromosoma' (dce/dce).
Fisher e Race si sono resi conto che gli 8 ‛cromosomi Rh' si presentavano in tre fasce di frequenza (v. fig. 9): molto frequenti, poco comuni e rarissimi (in modi diversi nei diversi gruppi razziali: per esempio, nei Negri il ‛cromosoma' più comune è il Dce); questi autori hanno allora suggerito che il primo gruppo sia costituito dai ‛cromosomi' originariamente presenti nella popolazione, mentre quelli del secondo gruppo vi si sarebbero accumulati nel corso di tempi relativamente lunghi anche in termini evolutivi (o meglio, microevolutivi) per crossing-over tra due dei ‛cromosomi' comuni. Il dCE, infine, sarebbe rarissimo perché il crossing-over necessario per produrlo dovrebbe essersi verificato in un eterozigote per uno dei ‛cromosomi' poco comuni. Secondo questa ipotesi, quindi, ‛cromosomi' contenenti d e C oppure d ed E oppure C ed E non esistevano nelle popolazioni da cui è derivata l'attuale popolazione inglese. Sempre secondo questa ipotesi: 1) un crossing-over, per mettere in cis C e d, deve verificarsi nella regione compresa tra questi due marcatori e in un individuo che possieda entrambi questi alleli, cioè in un eterozigote DCe/dce, la cui frequenza è circa 0,32; questo stesso crossing-over dà come prodotto complementare il ‛cromosoma Dce'; 2) analogamente, per produrre un ‛cromosoma dcE' e contemporaneamente, anche in questo caso, un Dce, si deve verificare un crossing-over tra d ed E in un eterozigote DcE/dce, la cui frequenza è circa 0,11; 3) infine, per produrre un ‛cromosoma DCE' occorre un crossing-over tra C ed E in un soggetto DCe/DcE, la cui frequenza è circa 0,11; anche in questo caso si produce contemporaneamente un Dce. La constatazione che la frequenza del ‛cromosoma Dce' è effettivamente all'incirca uguale alla somma delle frequenze dei ‛cromosomi dCe, dcE e DCE' è stata quindi un'importante conferma di questa ipotesi.
Queste considerazioni sono del tutto indipendenti dall'ordine dei tre marcatori nel ‛cromosoma Rh', dalle frequenze relative di dCe, dcE e DCE, e dalle frequenze degli eterozigoti nei quali questi ‛cromosomi' ricombinanti devono avere avuto origine. Ma, tenendo conto anche di questi ultimi dati, Fisher e Race hanno indicato la loro verosimile sequenza entro il ‛cromosoma Rh': infatti, è solo postulando che il marcatore Cc sia al centro che si rende conto in modo soddisfacente di questi dati quantitativi. Il fatto che le frequenze degli aplotipi ricombinanti dCe e dcE siano all'incirca uguali malgrado che l'opportunità di dare origine al primo (per crossing-over tra d e C) sia circa tre volte maggiore (0,32 e 0,11 rispettivamente) di quella di dare origine al secondo (per crossing-over tra d ed E), si spiega postulando che la distanza tra d e C sia circa tre volte minore di quella tra d ed E; d'altra parte il crossing-over tra C ed E appare essersi verificato molto di rado, e infatti DCE ha una frequenza circa quattro volte più piccola di quella di dcE, pur essendo circa uguali le opportunità di dare origine a questi due ‛cromosomi' ricombinanti; quindi la distanza tra C ed E è molto minore di quella tra d ed E. In conclusione, C è vicino agli altri due marcatori che appaiono lontani l'uno dall'altro, cioè C è in mezzo.
ζ) Un antigene X può dipendere da un effetto di posizione. Tali effetti sono messi in evidenza da antisieri che riconoscono antigeni associati non semplicemente ad alleli, ma ad assortimenti di alleli in cis: uno di essi, l'anti-c-e-, agglutina solo i globuli rossi di coloro che non solo possiedono almeno un allele c e uno e, ma li hanno ereditati dallo stesso genitore; un altro (l'anti- C-E-) agglutina solo quelli di coloro che hanno ereditato C ed E in cis; un altro ancora è specifico per C ed e in cis (anti- C-e-). Così, ad esempio, i globuli rossi dei soggetti DCE/dce sono agglutinati dai primi due di questi antisieri ma non dall'anti- C-e-, mentre quelli dei soggetti DCe/dcE, che hanno esattamente gli stessi alleli, assortiti però in modo diverso, sono agglutinati solo dall'anti- C-e-. I marcatori Cc ed Ee appartengono quindi, per definizione, allo stesso cistrone (si dice che due marcatori qualsiasi X e Y si trovano nello stesso cistrone se il fenotipo di X1Y1/X2Y2 è diverso da quello di X1Y2/X2Y1). Prima di questa scoperta avremmo potuto immaginare l'insieme dei tre marcatori strettamente associati che costituiscono il sistema Rh in due modi: come una serie di tre geni polimorfici diversi, anche se contigui, oppure come una serie di tre marcatori di una stessa unità funzionale, cioè di un singolo gene. La prima di queste due possibilità deve essere scartata: infatti, se i marcatori Cc ed Ee facessero parte di geni diversi, il fenotipo Rh dovrebbe dipendere solo dai singoli alleli presenti e non anche dal modo con cui sono assortiti.
È stato scoperto anche un effetto di posizione in trans: la quantità di antigene D che si trova nei globuli rossi di un soggetto con un gene D è ridotta quando un gene C si trova in trans rispetto a questo gene (i globuli rossi dCe/DCe hanno molto meno antigene D dei globuli rossi dce/DCe).
η) Un gene di gruppo sanguigno può interagire nella sua espressione con geni di altri sistemi. Questi sono forse i risultati che più si prestano a essere integrati e interpretati in termini biochimici. Anzi, talora essi hanno indicato la strada sulla quale le ricerche biochimiche dovevano procedere, come ad esempio nel caso del fenotipo Bombay (genotipo hh). L'analisi di varie famiglie con questo fenotipo eccezionale ha provato che tanto l'allele IA che quello IB del sistema AB0 richiedono, per esprimersi, il funzionamento di almeno un allele di un gene autosomico indipendente, detto H. Questo gene può esistere in due forme alleliche, quella funzionante, detta H, e una non funzionante, detta h, molto rara, per cui i soggetti hh sono assolutamente eccezionali. Questa relazione formale ha poi trovato la sua giustificazione quando si è dimostrato che gli antigeni A e B sono il prodotto finale di una catena biosintetica la cui tappa preterminale necessaria per l'espressione sierologica dei geni IA e IB è appunto determinata dal gene H (v. fig. 14).
Altri risultati classici a livello formale, anch'essi interpretati in termini di biosintesi di catene oligosaccaridiche con specificità antigeniche di gruppo sanguigno, sono le relazioni tra il sistema secretore (Se e se), il Lewis (L ed l), il gene Hh e l'AB0 (IA, IB e i).
Tra i risultati certamente di grande significato, ma che attendono ancora un'interpretazione molecolare, vanno almeno menzionati: un gene non identificato con un allele molto raro, trovato quindi solo allo stato eterozigote, che inibisce la comparsa degli antigeni di sistemi indipendenti tra loro come il Lutheran, il P e l'Auberger e forse anche altri; le relazioni tra sistemi Rh ed Lw; il fenotipo Rhnull, in genere, ma non sempre, associato ad anemia emolitica; alcune delle relazioni tra sistema AB0 e Ii; gli antisieri ‛eclettici', che riconoscono antigeni che dipendono dal genotipo per sistemi di gruppo sanguigno diversi, come l'antigene IB, l'IH, l'yTp1 e il Fy5 (in cui sembra coinvolta un'interazione tra i sistemi Duffy ed Rh). Anche se la natura di questi antigeni non è nota a livello molecolare, il solo fatto che esistano mostra come l'aver considerato la superficie dei globuli rossi come un mosaico di vari tipi di tessere completamente indipendenti (i vari antigeni di gruppo sanguigno) era solo una rappresentazione approssimativa della realtà. Le interazioni a livello biosintetico, come quelle dell'H, AB0 e Lewis, non sono più le sole interazioni immaginabili; è verosimile che ne esistano altre a carattere sterico tra molecole diverse e sintetizzate in modo indipendente. Ora che sappiamo che la membrana del globulo rosso è un mosaico liquido nel cui spessore possono spostarsi macromolecole con i determinanti antigenici a esse ancorate, la ricerca di altre interazioni è diventata possibile.
D. Bilancio dell'approccio sierologico. Esso è allo stesso tempo fortemente positivo, ma anche molto negativo.
I gruppi sanguigni rappresentano i migliori e quasi unici esempi di polimorfismi genetici per caratteri normali, e non solo per l'uomo; sono inoltre i migliori, se non gli unici, marcatori mendeliani di interesse antropologico, e hanno dato origine a quel ramo dell'antropologia, strettamente associato alla genetica di popolazioni, che si basa su frequenze geniche per caratteri alternativi invece che solo su caratteri antropometrici, i quali, essendo continui, sono in parte arbitrari e soggettivi (v. razza); sono stati inoltre i migliori marcatori genetici dell'uomo (tanto che per molto tempo negli unici tre esempi di linkage autosomici scoperti nell'uomo sono stati coinvolti sistemi di gruppo sanguigno) e gli unici marcatori genetici utilizzabili per l'accertamento della paternità. Essi hanno portato alla scoperta del linkage disequilibrium; hanno permesso di ipotizzare cooperazioni sequenziali tra geni nella biosintesi di macromolecole, aprendo e spianando la strada alla delucidazione della sintesi delle catene oligosaccaridiche che fungono da segnali sulla superficie cellulare; hanno fornito uno degli esempi più illustrativi di interazione tra polimorfismi (la protezione data dall'incompatibilità per il sistema AB0 verso le conseguenze dell'incompatibilità materno-fetale per il fattore Rh) e l'unico esempio ben documentato di polimorfismo universale e di vecchia data con svantaggio degli eterozigoti, anche se non di tutti (solo i feti Rh[+] figli di donne Rh[−] sono esposti a rischio); hanno infine risolto il problema delle trasfusioni e quello della malattia emolitica del neonato da fattore Rh. E tutto questo per più di cinquant'anni, cioè per circa due terzi dell'intera esistenza della genetica moderna, nata nel 1900 con la riscoperta delle leggi di Mendel. La sierologia - che ha così tanto contribuito allo sviluppo di tante altre scienze - per se stessa invece ha fatto molto meno. Essa infatti ha fallito in quello che si potrebbe considerare, almeno sul piano teorico, il suo compito principale, quello cioè di chiarire il significato biologico dei gruppi sanguigni, anzi addirittura nel rispondere al quesito pregiudiziale se abbiano o meno un significato. E, ciò che è ancora più grave, l'approccio sierologico è ‛intrinsecamente incapace' da solo di risolvere questo problema: non si tratta, cioè, di una questione di tempo, ma di principio. Per rendersene conto basta pensare che gli occhi del sierologo sono le cellule immunocompetenti degli individui che capitano alla sua osservazione e che queste cellule vedono, dei globuli rossi, solo ciò che si trova sulla loro superficie, e di questo solo ciò che differisce dai globuli rossi del loro stesso organismo (che, cioè, non è self). E tutto ciò in modo indipendente dalla sua quantità e dalla sua importanza: una struttura abbondante e fondamentale se è self non viene percepita, mentre una struttura non-self può costituire uno stimolo antigenico adeguato anche se la sua variazione è di piccola entità. Non appare azzardato, anzi, supporre che ci sia addirittura una relazione inversa: la natura stessa dei gruppi sanguigni - variazioni comuni tra individui normali - permette di escludere che queste variazioni abbiano un effetto fisiologico cospicuo. Naturalmente è possibile che i gruppi sanguigni abbiano un significato biologico, forse perfino importante, a livello evolutivo, ma su questo - come, del resto, sui polimorfismi enzimatici e proteici in genere, e sostanzialmente per gli stessi motivi - essendo le nostre conoscenze molto scarse, possiamo solo fare delle ipotesi. Ma per i gruppi sanguigni la nostra ignoranza va oltre questo livello comune di non spiegarci le variazioni individuali su base genetica: in questo caso, infatti, di regola non si conosce il significato nemmeno delle strutture stesse di cui si studiano le variazioni.
2. Livello biochimico. - I determinanti antigenici (o epitopi) di gruppo sanguigno sono, come accade per gli antigeni in generale, regioni relativamente piccole delle macromolecole di cui fanno parte che, a loro volta, non sono che una delle numerose componenti di una struttura ben più complessa, la membrana del globulo rosso.
Lo studio a livello biochimico di quasi tutti i sistemi di gruppo sanguigno in pratica deve essere ancora iniziato, e questo perché non si ha, o non si sa di avere, a disposizione il materiale di partenza, cioè la molecola antigenica isolata della membrana eritrocitaria.
Nell'affrontare lo studio della struttura e, ancor più, quello dei meccanismi genetici a livello molecolare che determinano la biosintesi degli antigeni eritrocitari, è necessario considerare separatamente due gruppi di sistemi: quelli i cui determinanti antigenici sono strutture oligosaccaridiche e quelli i cui determinanti antigenici sono parte di una catena polipeptidica. Per i primi la sintesi della struttura sierologicamente rilevante è il risultato dell'azione sequenziale di tante tappe biosintetiche quanti sono i monosaccaridi di cui essa è costituita. Per i secondi invece questa struttura dipende di regola da un solo gene, il gene strutturale della catena polipeptidica sede del determinante antigenico. I primi sono quindi situati alla fine o lungo una catena biosintetica, mentre i secondi richiedono la sintesi di una sola catena polipeptidica. Interazioni fenotipiche scoperte tra sistemi geneticamente indipendenti del primo gruppo (come, per esempio, il fatto che IA e IB non producono gli antigeni corrispondenti A e B in soggetti hh con il cosiddetto fenotipo Bombay: v. fig. 14) fanno pensare a meccanismi del tutto diversi da quelli che si devono ipotizzare per gli antigeni del secondo gruppo di sistemi.
Per i primi si deve ragionare in termini di interazioni tra enzimi: il prodotto di un enzima potrebbe costituire il substrato di un altro enzima (per es. enzima H ed enzimi A e B); oppure due enzimi potrebbero agire in modo diverso sullo stesso substrato, sullo stesso punto (per es. enzimi A e B sul residuo galattosilico terminale dell'antigene H) oppure in punti diversi di esso (per es. le fucosiltransferasi H ed L, che trasferiscono entrambe un residuo di fucosio, ma a residui diversi dello stesso accettore: v. fig. 14).
Interazioni genetiche che riguardino antigeni proteici devono invece far pensare a geni necessari non per la sintesi dell'antigene, bensì per uno degli eventi richiesti affinché il suo determinante risulti alla fine esposto sulla superficie della cellula e quindi identificabile a livello sierologico.
Quello che si vorrebbe conoscere di ogni antigene è prima di tutto la struttura del suo determinante e quella dell'intera molecola, quindi la sua esatta collocazione in rapporto alle altre molecole della membrana eritrocitaria, infine come si è giunti alla sua sintesi e quali sono i meccanismi che l'hanno portata e la mantengono dove si trova. A questo insieme di informazioni non si può certo giungere solo attraverso lo studio di antigeni isolati: occorre conoscere, oltre ai dati specifici sui singoli antigeni, anche quelli generali sulla struttura e la biosintesi delle membrane biologiche, in particolare di quella del globulo rosso. Solo così si può costruire il mosaico in cui inquadrare correttamente la tessera rappresentata da ogni singolo antigene.
Per poter discutere quello che si sa ora della biochimica dei gruppi sanguigni è necessario illustrare, sia pure in breve, la struttura della membrana eritrocitaria. Anche sotto questo aspetto il globulo rosso è la cellula eucariotica meglio conosciuta: esso è, infatti, non solo abbondante e facilmente disponibile, ma è anche l'unica ‛cellula' con una sola membrana, quella che la delimita al suo esterno.
Tutte le membrane biologiche, e quindi anche quella del globulo rosso, sono strutture lipidiche bistratificate e asimmetriche in cui sono sciolte proteine sintetizzate nel citoplasma (v. cellula: Fisiologia). Esse infatti consistono di due foglietti, ciascuno risultante da un insieme di molecole fosfo- o glicosfingolipidiche, cioè di molecole allungate tipicamente amfipatiche in quanto costituite da due regioni, una idrofilica e l'altra idrofobica (le due lunghe code idrofobiche ad andamento parallelo). Poiché tutte le molecole di uno stesso foglietto sono orientate nello stesso senso, il foglietto stesso presenta proprietà amfipatiche, cioè una superficie idrofilica e una idrofobica. Quest'ultima guarda la superficie idrofobica dell'altro foglietto: ne risulta una membrana con due superfici idrofiliche, di cui una citoplasmatica e l'altra che guarda all'esterno della cellula oppure nel lume di un organulo cellulare, separate da un contesto idrofobico il cui spessore, di poco più di una cinquantina di ångstrom, è pari al doppio della lunghezza delle code idrofobiche delle molecole (v. fig. 10).
La componente proteica di ogni membrana è formata da molecole ‛integrali' e ‛periferiche'. Le prime la attraversano a tutto spessore e quindi possono essere estratte solo con detergenti; le altre semplicemente aderiscono a una delle sue due superfici - di regola quella citoplasmatica - prendendo rapporto con un suo componente integrale.
La componente glucidica è costituita da sequenze oligosaccaridiche più o meno ramificate, ancorate con la loro estremità riducente a una proteina integrale (formando cosi una glicoproteina) o a un lipide, detto cerammide (formando così un glicosfingolipide), costituito da un amminoalcool, la sfingosina, e da un acido grasso che è in genere l'acido stearico (v. fig. 11). La parte glucidica dei glicosfingolipidi è legata all'estremità idrofilica del cerammide, mentre le due code idrofobiche di quest'ultimo corrispondono una alla sezione idrofobica della sfingosina e l'altra all'acido grasso, che è l'acido stearico (v. fig. 11).
Una delle caratteristiche principali di qualsiasi membrana biologica è, come si può vedere nella fig. 10, l'elevato grado di asimmetria esistente tra le sue due facce. Esso riguarda soprattutto la componente glucidica, che è rivolta solo all'esterno, e quella proteica: per ogni tipo di proteina le molecole presentano tutte lo stesso orientamento. I due foglietti però sono alquanto diversi anche per ciò che riguarda la loro composizione lipidica.
Per collocare al posto giusto una certa sostanza (per esempio una glicoproteina sulla superficie esterna della membrana cellulare) la cellula deve sintetizzare la catena polipeptidica, ancorare a essa le catene oligosaccaridiche con le loro sequenze specifiche, far giungere questa molecola a ‛destinazione' e imporle il corretto orientamento (cioè con la sezione a cui sono ancorate le catene oligosaccaridiche rivolta all'esterno e sporgente al di fuori della cellula).
Non tutti i meccanismi che portano a questo risultato finale sono stati compresi, ma un quadro corretto si è ormai riusciti a tracciarlo. Si tratta di uno dei progressi più significativi della biologia da quando si è arrivati a comprendere il controllo genetico della struttura delle proteine. Infatti è solo seguendo questa strada che si può arrivare a comprendere la morfogenesi delle strutture sopramolecolari e, in definitiva, il controllo genetico della struttura della cellula come insieme integrato, invece che solo delle singole proteine in essa contenute.
Un primo punto fondamentale è che il gene strutturale di ogni catena polipeptidica ne determina non solo la sequenza amminoacidica e, attraverso di essa, l'architettura tridimensionale, ma anche il destino topologico (sede e orientamento), in quanto anch'esso è diretta conseguenza della struttura primaria della proteina. La sintesi di tutti i polipeptidi inizia su un ribosoma libero nel citoplasma, ma le catene destinate ad attraversare una membrana (per essere poi secrete o situarsi nel lume di un organello cellulare) oppure a rimanervi ancorate come proteine integrali, hanno di regola una sequenza di inizio molto idrofobica, detta ‛sequenza segnale', che permette loro di traversare a tutto spessore una membrana del reticolo endoplasmatico mentre la catena polipeptidica è ancora allo stato ‛nascente'. La successiva crescita sequenziale della catena polipeptidica prosegue secondo le solite regole, seguendo l'informazione contenuta nella molecola di mRNA rimasta all'esterno e legata al ribosoma sul quale si sta effettuando la sintesi. La sequenza segnale viene rimossa ancora prima del completamento della sintesi della catena polipeptidica (per questo la si è scoperta tanto in ritardo); alla fine della sintesi la catena polipeptidica, grazie alla sua sequenza iniziale ormai rimossa, si trova ad aver superato almeno in parte la barriera, altrimenti insormontabile, costituita dal doppio foglietto idrofobico della membrana del reticolo endoplasmatico. Se si limiterà ad affacciarsi nel lume di questo organello - come accade se, oltre a quella segnale, esiste nella sua sequenza (cioè, in ultima analisi, nel suo gene strutturale) un altro lungo tratto idrofobico che la tiene ancorata alla membrana - oppure si troverà libera al suo interno - come nel caso delle proteine destinate a essere secrete - anche questo è scritto, sia pure indirettamente, nel suo gene strutturale. Poiché il lume del reticolo endoplasmatico ha, similmente ad altri organelli cellulari delimitati da una membrana come l'apparato di Golgi e i lisosomi, le proprietà topologiche e strutturali di un ‛esterno internalizzato', il risultato raggiunto è che le proteine che iniziano con una ‛sequenza segnale' sono di fatto esterne, dato che possono affacciarsi all'esterno della cellula o essere secrete, a seconda dei casi, senza dover attraversare la membrana cellulare. È sufficiente, affinché questo accada, che una vescicola il cui lume sia un ‛esterno internalizzato' si fonda con questa membrana; questo processo non richiede un'inversione di orientamento delle strutture della membrana della vescicola, in quanto dei due foglietti della vescicola quello che guarda verso il suo lume e che in seguito alla fusione con la membrana cellulare si troverà a guardare all'esterno della cellula ha la stessa struttura del foglietto esterno della membrana cellulare (per esempio presenta strutture oligosaccaridiche); lo stesso vale per il foglietto esterno della vescicola che è bagnato dal citoplasma come il foglietto interno della membrana cellulare. È questo il senso del concetto di ‛esterno internalizzato' proposto per la prima volta da Palade (v. figg. 10 e 12). In conclusione, l'ultima fase del destino di una proteina ‛esterna' - il suo trovarsi all'‛esterno' della cellula che l'ha prodotta - è in effetti, dal punto di vista topologico, il primo evento della sua sintesi.
A sintesi proteica ultimata, prima che la molecola proteica si affacci effettivamente sulla superficie esterna della cellula o che pervenga nel lume di un organello cellulare oppure ancora che sia secreta, si svolge - sempre all'interno di vescicole endoplasmatiche - tutto l'insieme dei processi che culminano con l'addizione a questa molecola delle sue appendici oligosaccaridiche. L'inizio di questo processo, cioè la formazione del tratto prossimale delle catene oligosaccaridiche, dipende dalla natura della molecola a cui devono essere attaccate queste appendici: si può trattare di una proteina destinata a diventare una glicoproteina, oppure di una molecola di cerammide destinata a diventare un glicosfingolipide. Ma superata questa prima fase, il seguito del processo, fino al raggiungimento delle sequenze finali delle varie catene oligosaccaridiche, è verosimilmente lo stesso nei due casi. Le figg. 13 e 14, che si riferiscono a una glicoproteina e a un glicosfingolipide, possono quindi servire da riferimento per entrambe le molecole, purché si prenda in esame l'accrescimento delle sole parti distali dei vari oligosaccaridi che vi sono rappresentati.
L'aspetto più interessante, ma anche più difficile da chiarire, della sintesi delle catene oligosaccaridiche è che, per generare le loro sequenze, la cellula non dispone di una molecola stampo, come avviene in modo diretto nella trascrizione degli acidi nucleici e in modo indiretto nella traduzione delle proteine, che sono inoltre sequenze di unità unite tutte con un unico tipo di legame, quello fosfoesterico 5′-3′ dei polinucleotidi e quello peptidico delle proteine. Invece, nel caso delle sequenze saccaridiche, va specificata non solo la sequenza dei monosaccaridi, ma anche la direzione del legame rispetto al ‛piano' del monosaccaride aggiunto alla catena (α o β) e la posizione dell'atomo di carbonio dello zucchero accettore che viene impegnato in questo legame con l'atomo di carbonio riducente (l'1) del monoso aggiunto (1→2 o 1→3 o 1→4). Infine, le catene oligosaccaridiche possono essere - e anzi spesso sono - ramificate se non addirittura ‛rigogliosamente fronzute', cioè sono suscettibili di crescere in più punti contemporaneamente, a differenza delle catene polinucleotidiche e polipeptidiche. Solo un enzima può essere responsabile di una reazione di cui debba essere specificata una serie così grande di variabili; quindi ogni singola addizione a un oligosaccaride che abbia una sequenza specifica implica l'esistenza di un enzima, il che significa che per la sintesi dell'intera sequenza devono essere implicati vari enzimi. È facile però rendersi conto che questo ancora non basterebbe: se tutti questi enzimi potessero agire contemporaneamente su un miscuglio disordinato di catene oligosaccaridiche nascenti, che si trovassero cioè nelle fasi più svariate del loro accrescimento, non ne risulterebbe la produzione di sequenze relativamente omogenee, almeno nella loro parte iniziale, come invece si verifica, oppure la loro sintesi sarebbe poco efficiente, dato che molti enzimi dovrebbero restare inattivi in attesa della sintesi del loro substrato da parte degli enzimi che li devono precedere. Si è quindi postulato che l'accrescimento di queste catene sia non solo un processo sequenziale che procede con l'addizione di un monosaccaride alla volta (come è stato effettivamente dimostrato), ma che i vari enzimi responsabili dell'accrescimento di una data catena oligosaccaridica siano disposti come in una catena di montaggio lungo il tragitto che deve percorrere la molecola con la sua catena oligosaccaridica nascente, come è mostrato nella fig. 13. Questo comporta che una catena oligosaccaridica, che abbia superato la regione del canalicolo sede di una certa glicosiltransferasi senza aver subito l'aggiunta del monoso trasferito da quell'enzima, non è più suscettibile di subire quel tipo di apposizione e quindi nemmeno di andare incontro al tipo di accrescimento per il quale questa apposizione è necessaria. Questo spiega perché di regola i vari determinanti antigenici oligosaccaridici si trovano insieme ad altri che ne sono di fatto i precursori: evidentemente l'accrescimento di ogni catena oligosaccaridica può arrestarsi a qualsiasi livello intermedio, oltre a poter imboccare in certi punti vie biosintetiche alternative (v. figg. 13 e 14). E poiché ciascuna delle catene diverse della stessa o di differenti molecole incontra a ogni passo della sua crescita sequenziale queste varie possibilità, le formule dei mucopolisaccaridi hanno un valore statistico: un mucopolisaccaride purificato non è un insieme di molecole identiche bensì una famiglia di molecole con strutture, e perfino formule brute, distribuite attorno a valori medi, ciascuna delle quali è sede di numerosi determinanti antigenici, alcuni uguali e alcuni diversi tra loro (per es., i determinanti A e B dei mucopolisaccaridi dei secretori di gruppo AB coesistono nelle stesse molecole) originandosi così un'estrema eterogeneità sia intra- (cioè tra catene oligosaccaridiche legate alla stessa catena polipeptidica), sia intermolecolare nell'ambito di ogni sostanza mucopolisaccaridica ‛pura'. Tale eterogeneità, tuttavia, non è responsabile dell'eterogeneità dei gruppi sanguigni: le variazioni individuali a cui essa può dar luogo sono solo di tipo quantitativo, mai addirittura alternative come quelle responsabili delle differenze tra antigeni di gruppo sanguigno.
In questo tipo di biosintesi sequenziale, che richiede che le varie parti unitarie della catena siano allineate e saldate in modo specifico, le unità di informazione che devono essere disposte in modo co-lineare rispetto alla catena oligosaccaridica sono gli enzimi specifici per ogni apposizione: i codoni di ogni catena oligosaccaridica sono i vari enzimi glicosiltransferasici che si succedono lungo il canalicolo endoplasmatico; quest'ultimo corrisponde a un ribosoma.
Gli ordini di grandezza dell'apparato richiesto per sintetizzare una macromolecola, seguendo le istruzioni contenute nella corrispondente struttura che ne costituisce il programma, sono le decine di ångstrom per gli acidi nucleici per i quali questo apparato si riduce in sostanza a un complesso enzimatico; le centinaia di ångstrom per le catene polipeptidiche (apparato poliribosomico); le migliaia di ångstrom, fino ad arrivare a strutture al limite di risoluzione addirittura del microscopio ottico, per le catene oligosaccaridiche, in cui ogni unità informazionale ha una complessità paragonabile a quella dell'intero apparato che provvede alla sintesi degli acidi nucleici.
3. Integrazione tra genetica formale e biochimica dei gruppi sanguigni. - Una trattazione sistematica dei gruppi sanguigni esula completamente dagli scopi di questo articolo, quindi ci limiteremo a presentare solo il minimo di dati indispensabile a illustrare ‛lo stato attuale dell'arte', cioè il livello raggiunto in vari sistemi o gruppi di sistemi nell'integrare fra loro due linee di ricerca e di comprensione differenti: quella della genetica formale (a cui si è arrivati con tecniche sierologiche) e quella biochimica (frutto dell'analisi chimica degli antigeni solubili, prima, e di quelli estratti dalle membrane eritrocitarie, in seguito).
L'integrazione tra questi due tipi di dati è spesso difficile, perché può anche capitare che un certo antigene sia noto a livello genetico e sierologico e sia stato anche caratterizzato più o meno completamente a livello biochimico (per esempio come banda elettroforetica in un protidogramma delle proteine estratte dalla membrana eritrocitaria), ma che nessuno si sia accorto di questa identità.
Due sono i fattori biologici da cui maggiormente è dipeso il grado di comprensione integrata genetico-biochimica dei vari sistemi di antigeni: a) l'esistenza o meno di questi antigeni anche in forma idrosolubile nelle secrezioni e nei liquidi biologici in genere; b) la loro abbondanza. Non è certo per caso che il gruppo di sistemi meglio compresi dal punto di vista genetico e biochimico siano l'AB0 e i sistemi che interagiscono con esso, i cui antigeni esistono anche (o prevalentemente) come mucopolisaccaridi solubili e si trovano tanto nelle secrezioni che nella membrana eritrocitaria in grandi quantità (dell'ordine di un milione di determinanti antigenici per globulo rosso, in confronto ai 50-100.000 del sistema MN, ai circa 10.000 dell'Rhe a cifre ancora più basse per gli altri sistemi).
A. Sistemi degli antigeni AB0, Lewis, secretore e Hh (v. fig. 14). I primi tre di questi geni sono polimorfici: esistono tre alleli comuni, IA (a sua volta suddiviso in IA e IA2), IB e i, nel sistema AB0; due alleli comuni, L ed l, nel sistema Lewis; e due alleli comuni, Se ed se, nel sistema secretore. L'ultimo sistema non è polimorfico, perché dei suoi due alleli, H e h, il secondo è molto raro, per cui gli omozigoti hh sono assolutamente eccezionali.
Il gene Hh è il gene della fucosiltransferasi che genera il determinante antigenico H: l'allele H produce questo enzima negli eritroblasti, qualunque sia il genotipo dell'individuo per il gene secretore, mentre la sua espressione nelle secrezioni (come presenza sia di questa attività enzimatica, sia dell'antigene H da essa prodotto) richiede la presenza di almeno un gene Se. I rarissimi soggetti hh non producono in nessun caso antigene H né negli eritrociti né nelle secrezioni.
L'allele L del sistema Lewis, che è presente almeno in singola dose in circa il 90% dei Caucasici, produce una fucosiltransferasi diversa dalla precedente, in quanto agisce solo su glicoproteine di secrezione (e quindi non produce né contribuisce a produrre antigeni eritrocitari autoctoni) e trasferisce il fucosio alla N-acetilgalattosammina subterminale della catena oligosaccaridica invece che al suo galattosio terminale, generando così il determinante antigenico Lea invece del determinante antigenico H. Quando entrambi questi enzimi agiscono sulla stessa catena si genera il determinante antigenico Leb. I soggetti ll non producono questa fucosiltransferasi e quindi non possiedono né l'antigene Lea né l'Leb.
Il gene AB0, infine, consta di tre alleli di cui uno, l'i, è almeno apparentemente un allele muto, al quale non è associata alcuna attività glicosiltransferasica; gli altri due producono invece due differenti glicosiltransferasi: l'IA produce una N-acetilgalattosamminiltransferasi capace di legare la N-acetilgalattosammina al galattosio di una catena oligosaccaridica, purché a esso sia legato un residuo di fucosio a opera del gene H; l'IB produce una galattosiltransferasi che ha lo stesso accettore dell'enzima precedente. Il trasferimento della N-acetilgalattosammina genera il determinante antigenico A e quello del galattosio il determinante B; in entrambi i casi i determinanti antigenici preesistenti (l'H oppure l'Leb) vengono mascherati.
È ora possibile considerare brevemente le condizioni genotipiche necessarie alla comparsa dei determinanti antigenici di questi sistemi nelle secrezioni e nei globuli rossi.
La presenza degli antigeni A e B nella membrana entrocitaria dipende in pratica solo dalla presenza degli alleli IA e IB rispettivamente, visto che il prerequisito della presenza dell'antigene H è sempre soddisfatto (eccetto che nei rarissimi individui hh, con fenotipo Bombay). Per quanto riguarda le secrezioni, invece, la presenza di IA e di IB non implica necessariamente la presenza degli antigeni corrispondenti, perché una considerevole proporzione di individui (circa il 25% tra i Caucasici) sono sese, non possiedono cioè nemmeno un allele Se che è necessario almeno in singola dose all'espressione del gene H, a sua volta necessaria all'espressione dei geni IA e IB. Quindi la produzione dei determinanti A e B dal loro precursore richiede la cooperazione di due geni (almeno un H più un IA e/o un IB) se si tratta di globuli rossi, e di tre geni (gli stessi di prima più almeno un Se) se si tratta di antigeni solubili.
I globuli rossi possono diventare Le(a+) o Le(b+), cioè agglutinabili rispettivamente dai sieri anti-Lea o antiLeb, solo se questi antigeni raggiungono nel plasma la concentrazione sufficiente a ‛verniciarli' a dovere. Se si escludono gli ll che non producono nessuno di questi due antigeni, il fenotipo Lewis salivare e plasmatico e quindi anche eritrocitario dipende da quello che fa il gene H (che è sempre presente): se esso non funziona perché il soggetto in questione non ha nemmeno un gene Se, tutto il determinante Lea prodotto dall'enzima L rimane tale e allora ve ne è molto nella saliva e anche nel plasma, per cui i globuli rossi diventano Le(a+); se invece il gene H funziona perché c e almeno un gene Se, gran parte delle catene carboidratiche acquistano una specificità Leb, per cui c'è abbastanza Leb da rendere Le(b+) i globuli rossi, ma non resta Lea a sufficienza per renderli Le(a+): quest'ultimo antigene è presente, anche se in quantità modesta, nella saliva, ma i globuli rossi risultano Le(a−). Quindi si possono trovare soggetti Le(a−b−) (gli ll), ma non soggetti Le(a+ b+): questi due antigeni, nei globuli rossi, si escludono a vicenda. In conclusione, i fenotipi Lewis risultano dalla cooperazione di ben tre geni, due dei quali (Ll e Sese) polimorfici: per sintetizzare l'antigene Leb devono lavorare sullo stesso moncone oligosaccaridico tanto l'enzima L che l'H (condizione quest'ultima che richiede la presenza di almeno un gene Se e che quindi non viene soddisfatta in tutti i non secretori sese, che sono molti); per sintetizzare antigene Lea basta il solo gene L, ma perché ne rimanga tanto da rendere Le(a+) i globuli rossi occorre che il gene H non funzioni nelle secrezioni, cioè che il genotipo del soggetto sia sese. In altre parole, nell'ambito degli individui con almeno un gene L (circa il 9o% dei Caucasici) sono Le(a+) i soggetti sese ed Le(a−) gli SeSe e gli Sese: questo antigene si comporta quindi come un carattere recessivo perché richiede l'omozigosi per un allele che però non è un allele del gene L, cioè del gene che lo produce (il che sarebbe risultato incomprensibile), bensì di un altro gene che controlla l'espressione di un enzima che agisce sullo stesso substrato su cui agisce l'enzima L.
Questi sistemi sono i più adatti a illustrare le complesse interazioni che possono verificarsi nel caso che geni di gruppo sanguigno codifichino non per gli antigeni stessi, bensì per gli enzimi responsabili della sintesi di questi antigeni. Essi offrono inoltre l'esempio più completo attualmente noto di integrazione tra dati genetici e dati biochimici: l'analisi formale della trasmissione ereditaria di questi caratteri aveva portato a risultati sicuri, per molti dei quali però era difficile immaginare - si badi bene: ‛immaginare' - una spiegazione plausibile. La genetica del sistema Lewis, in particolare, con un antigene trasmesso come carattere recessivo solo a livello eritrocitario e non in tutte le persone, avrebbe continuato a costituire una stranezza della natura, oltre a rappresentare un notevole successo dei genetisti (Grubb e Ceppellini) che erano riusciti a chiarirla a dispetto della sua complessità, se la delucidazione della struttura dei determinanti antigenici H, Lea ed Leb, prima, e l'identificazione degli enzimi H ed L responsabili, in seguito, non ne avesse fornito la logica spiegazione.
Naturalmente non tutto è ancora chiarito di questi sistemi. Restano aperti, tanto per cominciare, due grossi problemi: la natura e la funzione del gene Sese (si sa solo che il gene H si esprime sui mucopolisaccaridi delle secrezioni solo se è presente almeno un allele Se) e la natura della differenza tra l'enzima A e l'enzima B, tanto a livello proteico (quanto sono diversi questi due enzimi che trasferiscono allo stesso accettore esclusivamente la N-acetilgalattosammina o esclusivamente il galattosio?) che a livello genetico (sono veramente allelici questi due geni che producono due enzimi che catalizzano reazioni diverse? Se è così, si tratta di una situazione biologica a dir poco inconsueta).
B. Sistema degli antigeni P, P1 e Pk (v. fig. 14). Il livello delle conoscenze attuali sulla biosintesi e la genetica dei determinanti di questi antigeni non è così avanzato come quello dei sistemi precedenti: si conoscono ormai le loro formule di struttura, ma non gli enzimi che li producono né, con sicurezza, le relazioni tra i geni che producono questi enzimi. Salvo individui eccezionali (omozigoti per un allele raro), nei globuli rossi non si trova mai l'antigene Pk e si trova sempre l'antigene P. Il contrario si verifica nei fibroblasti. Ciò accade perché tutti, anzi quasi tutti, gli individui hanno almeno un allele per la β(1→3)-acetilgalattosamminiltransferasi, cioè per l'enzima che trasforma l'antigene Pk in antigene P, ma questo gene si esprime nelle cellule delle serie rossa e non in altre cellule come i fibroblasti. L'antigene P rientra quindi nel gruppo degli antigeni a cui appartengono quasi tutti gli antigeni di gruppo sanguigno, che si trovano solo nella membrana dei globuli rossi. È presumibile, tuttavia, che non si tratti, come potrebbe sembrare, di un antigene di differenziazione (cioè di un antigene necessario alla differenziazione degli eritrociti), bensì semplicemente di un antigene distribuito in modo differente in cellule che si sono differenziate con meccanismi che non richiedono la sua presenza. Infatti i sia pure rarissimi individui con antigene Pk e senza antigene P nei loro globuli rossi non presentano, per quanto ne sappiamo, alcuna alterazione eritrocitaria.
La relazione biosintetica Pk→P spiega come mai la presenza dell'antigene Pk negli eritrociti si comporti come un carattere recessivo: affinché l'antigene Pk rimanga, invece di essere trasformato in P, è necessario che manchi l'attività enzimatica responsabile di questa trasformazione, il che accade solo negli omozigoti per un allele silente del gene che produce questa transferasi.
C. Sistema MNS. Questo sistema consiste di almeno due geni, l'MN e l'Ss, così strettamente concatenati da trovarsi in linkage disequilibrium (tra i Caucasici, per esempio, l'aplotipo o ‛cromosoma MS' è più frequente del prodotto della frequenza dell'allele M per la frequenza dell'allele S). Sembra che l'MN e l'Ss siano i geni strutturali della glicoforina A (la principale glicoproteina della membrana del globulo rosso) e, rispettivamente, della B.
La glicoforina A è la glicoproteina meglio conosciuta di tutte le glicoproteine delle membrane cellulari. È costituita da una catena polipeptidica di 131 residui amminoacidici, di cui si conosce la sequenza completa, e da 16 catene oligosaccaridiche tutte legate al primo tratto, cioè a quello che comincia con il residuo terminale NH2, che è lungo 64 residui amminoacidici. Questo primo tratto idrofilico, che pesca nel plasma in cui sono immersi i globuli rossi, contiene appunto 16 residui a cui sono legate altrettante catene oligosaccaridiche. A esso segue una sezione fortemente idrofobica di 32 residui che attraversa a tutto spessore la membrana eritrocitaria trovandosi quindi immersa nella matrice idrofobica del doppio foglietto fosfolipidico. L'ultimo tratto, di nuovo idrofilico, di 35 residui amminoacidici è la sezione COOH-terminale della glicoforina.
La glicoforina A isolata dai soggetti M differisce in due punti della sua sequenza iniziale da quella dei soggetti N (M=H2N-‛Ser', Ser, Thr, Thr, ‛Gly'; N=H2N-‛Leu', Ser, Thr, Thr, ‛Glu'), ma per vari motivi (tra cui il fatto che il residuo Ser presente nella glicoforina M e assente nella N è proprio uno di quelli da cui si originano catene oligosaccaridiche) sembra che la base strutturale della differenza sierologica tra gli antigeni M ed N non sia la diversità delle sequenze amminoacidiche delle rispettive glicoforine o, perlomeno, che questa non sia la sola differenza. Si ritiene invece che la componente oligosaccaridica contribuisca in modo rilevante al comportamento antigenico, M o N, dell'intera molecola. È comunque verosimile che la struttura della componente carboidratica di questa molecola dipenda dalla sequenza amminoacidica della sua componente proteica. Se è così, allora l'MN è effettivamente il gene strutturale della glicoforina.
In ogni caso il gene della glicoforina A presenta l'esempio meglio conosciuto a livello molecolare di linkage disequilibrium tra due siti dello stesso gene strutturale, il suo primo e il suo quinto codone, entrambi polimorfici nelle stesse popolazioni. Si tratta di un disequilibrio praticamente assoluto, dato che, dei due possibili aplotipi ricombinanti, uno (l'Mc, la cui sequenza è ‛Ser', Ser, Thr, Thr, ‛Glu') è estremamente raro e l'altro (che dovrebbe avere la sequenza ‛Leu', Ser, Thr, Thr, ‛Gly') non è stato mai trovato. È ragionevole supporre che il fatto che quasi non esistano aplotipi ricombinanti sia da attribuirsi alla vicinanza tra i due siti polimorfici (se - come è verosimile - tra di essi non si interpone alcun introne, la distanza che li separa è di solo dodici coppie di desossiribonucleotidi). Quindi a rigore M ed N sono aplotipi piuttosto che alleli. Essi, a loro volta, sono in linkage disequilibrium con gli alleli S ed s di un gene diverso ma molto vicino, quello della glicoforina B.
Della glicoforina B, sede dei determinanti antigenici S ed s, si sa molto meno. Un dato interessante è che essa presenta, anche se debolmente, una certa antigenicità di tipo N, e questo spiega la presenza di una lieve specificità N anche nei soggetti MM. In generale esistono due alleli di questo gene, l'S e l's, che producono gli antigeni omonimi, ma nei Negri è molto comune un altro allele, l'Su che non produce né l'uno né l'altro di questi due antigeni e che potrebbe anche essere un gene silente (glicoforina B-).
Nel complesso, sebbene la struttura della glicoforina A sia completamente nota, la struttura della glicoforina B e, più in generale, quella dei determinanti antigenici del sistema MNS rimangono ancora da chiarire.
D. Sistema Rh. Le conoscenze genetiche su questo sistema sono giunte a un grado estremo di sofisticazione e di complessità. È certamente coinvolto un numero molto grande di alleli diversi dal punto di vista qualitativo e non, come nel sistema AB0, essenzialmente sul piano quantitativo. Questo presumibilmente dipende almeno in parte dal fatto che, trattandosi di antigeni di natura proteica, il numero di possibili prodotti allelici qualitativamente diversi l'uno dall'altro è molto grande; nel caso di determinanti antigenici di natura oligosaccaridica, invece, una differenza allelica qualitativa è generalmente del tipo presenza-assenza di un certo monoso in una certa posizione della catena (allele a cui è associata un'attività glicosiltransferasica e allele a cui non è associata questa attività: H invece che h, L invece che l, ecc.) e solo eccezionalmente esistono due alleli a cui sono associate glicosiltransferasi diverse, come si verifica per gli alleli IA e IB, sempre ammesso che siano veramente alleli. È comunque certo che non esistono intere serie alleliche di enzimi che catalizzano reazioni qualitativamente diverse.
Il risultato più importante, oltre che del tutto inatteso, delle ricerche biochimiche su questo sistema è stato la scoperta che dalle membrane di globuli rossi Rh(−), cioè D(−), si può estrarre la stessa quantità di antigene D che dalle membrane dei globuli rossi Rh(+). In altre parole, la Rh negatività non è dovuta all'assenza di antigene Rh, bensì alla sua non esposizione in senso sierologico (i globuli rossi Rh negativi, oltre a non essere agglutinati dai sieri anti-Rh, per definizione, non inducono la comparsa di anticorpi anti-Rh in animali opportunamente immunizzati), il che in prima approssimazione equivale a dire che i globuli rossi Rh negativi contengono sì antigene Rh, ma che esso non guarda all'esterno. È probabile che si tratti di una differenza strutturale tra molecole proteiche alleliche che ne influenzi l'orientamento rispetto alla membrana in cui sono situate. Questo sarebbe allora, se non il primo, certo uno dei primi esempi di mutazione a effetto topologico, riguardante, cioè, una parte della molecola proteica da cui dipendono la sua localizzazione nella cellula e il suo orientamento.
E. Sistemi Chido (Ch) e Rodgers (Rg). Sono gli unici sistemi di gruppo sanguigno situati nella regione HLA. Questi due antigeni rivestono un grande interesse, perché - caso unico per i gruppi sanguigni - se ne conosce la funzione: essi costituiscono la componente C4 del complemento. Si tratta cioè di sostanze originariamente plasmatiche che, come gli antigeni Lea ed Leb, sono adsorbite alla superficie esterna dei globuli rossi a cui conferiscono le proprietà antigeniche corrispondenti (v. immunologia e immunopatologia: Immunologia generale e Malattie autoimmuni).
Sembra ormai accertato che il C4 dipenda da due geni strutturali (situati, come tutti i geni del complemento, nella regione HLA) ciascuno con due alleli comuni, di cui uno silente e quindi recessivo, e uno che produce anche in singola dose una proteina C4 riconoscibile sia nel plasma, con una tecnica immunoelettroforetica, sia nei globuli rossi, con una prova di agglutinazione. Il fenotipo C4(−) (cioè l'omozigote per l'aplotipo con gli alleli silenti di entrambi i geni C4) è molto raro, perché questi alleli silenti non si trovano quasi mai in cis. È possibile che al mantenimento di questo disequilibrio abbia contribuito anche la selezione, dato che l'aplotipo con entrambi i geni silenti è certamente svantaggioso allo stato omozigote.
4. Perché esistono le sostanze di gruppo sanguigno? Perché esistono i gruppi sanguigni? - Come dice il loro stesso nome, le sostanze ‛di gruppo sanguigno' presentano variazioni individuali che sono, almeno in alcuni casi, l'unico elemento che permette di identificarle. Si tratta, cioè, di sostanze di cui scopriamo l'esistenza solo nel caso che se ne scopra una variazione individuale. Non possiamo identificarle con un nome che ne indichi la natura chimica o la funzione (come enzimi, o proteine di rivestimento, o recettori, ecc.), perché non conosciamo né l'una né l'altra. L'unica cosa nota è che variano, ma spesso non sappiamo né in che consista la variazione né che tipo di molecola sia quella che presenta questa variazione.
Questo stato di cose, che si estrinseca necessariamente già al livello del loro nome (sostanze di gruppo è come dire: sostanze che variano), ha spesso ingenerato un equivoco; le sostanze di gruppo sanguigno, infatti, sono state spesso considerate un po' alla stregua di ‛distintivi', quasi che la loro funzione fosse quella di variare da un individuo all'altro, mentre la variazione è solo ciò che permette a noi di scoprirle, non ciò che permette loro di funzionare. In altre parole, occorre non lasciarsi influenzare dal fatto che queste sostanze sono state scoperte grazie alla loro variabilità.
È quindi evidente che le domande che ci siamo poste non sono formulate in modo corretto, ma appunto in questa forma hanno costituito per decenni uno degli argomenti più dibattuti della genetica.
È chiaro che vi sono buone probabilità di trovare una risposta a tali questiti solo se riguardano le sostanze di cui sia ormai nota la struttura chimica. In altri termini, se può ora aver senso chiedersi a cosa servono per esempio le molecole AB0, cioè quelle date catene oligosaccaridiche che sono legate con legame glicosidico agli sfingolipidi della membrana eritrocitaria, continua a non avere interesse biologico domandarsi a cosa serve, per esempio, la sostanza del sistema Kell di cui non conosciamo la natura.
È comunque assai poco verosimile che i vari tipi di molecole della membrana eritrocitaria che possono essere sede di determinanti antigenici di gruppo abbiano in comune qualche importante proprietà, a parte quella di essere esposte all'esterno per cui una loro eventuale variazione è scopribile dal sistema immunitario.
Il fatto di conoscere una grande variabilità della superficie esterna degli eritrociti si può ragionevolmente spiegare - senza ricorrere a ipotesi speciali rispetto a quelle sulla variabilità genetica in generale - tenendo conto che: a) questa variabilità non è che il sottoprodotto di una caratteristica ben più rilevante della variabilità stessa, cioè che la superficie cellulare, invece di essere costituita solo dall'insieme delle estremità polari dei lipidi di membrana (strutture troppo semplici per poter variare), è costellata da un gran numero di strutture molto più complesse, capaci quindi, oltre che di svolgere funzioni specifiche, anche di presentare variazioni di ‛gruppo'; b) il sistema immunitario è carattenzzato da una grande ‛bravura' nello scoprire differenze individuali; c) è stato effettuato un enorme numero di prove di compatibilità trasfusionale, ciascuna delle quali ha di fatto costituito l'equivalente di una deliberata ed efficientissima ricerca di una variabilità di gruppo.
Per alcune sostanze, almeno per quanto ci risulta, la variabilità può essere anche del tipo presenza-assenza, il che implica che la loro funzione - visto che si tratta di variazioni individuali non patologiche - non è indispensabile (o perché la funzione è in sé non necessaria o perché può essere svolta anche da un altro tipo di molecole). Questo è forse il caso dell'antigene Duffy che manca tanto nella sua forma a che in quella b (e che quindi forse è effettivamente assente negli omozigoti FyFy molto frequenti tra i Negri). Poiché questi soggetti sono assolutamente immuni dalla malaria da Plasmodium vivax, si pensa che questa molecola sia il recettore eritrocitario di questo plasmodio (v. anche razza). Se è così, il fatto che non si sia diffusa una resistenza nei confronti di Plasmodium falciparum su base analoga, cioè attraverso la perdita del suo recettore, il che avrebbe costituito un vantaggio di gran lunga maggiore, suggerisce che questo ipotetico recettore sia indispensabile: l'essersi scelto un recettore irrinunciabile sarebbe un bell'esempio di ‛astuzia evolutiva' di Plasmodium falciparum rispetto a Plasmodium vivax.
Il significato funzionale di una variazione di gruppo per un determinante antigenico dipende fortemente dalla sua natura chimica: se esso è di natura proteica, è presumibile che la variazione consista nel fatto che in due proteine alleliche, per il resto identiche, un residuo amminoacidico è sostituito da un altro; se invece si tratta di un determinante carboidratico, allora una differenza qualitativa implica l'esistenza di un enzima al posto di un altro.
Le funzioni delle molecole antigeniche, anche di quelle di cui ormai si conosce la natura chimica, sono tuttora in massima parte sconosciute, tanto che si potrebbe addirittura dubitare che esse abbiano effettivamente una funzione. Tuttavia, pur mancando una prova assoluta di un loro significato funzionale, non è ragionevole supporre che una intera catena biosintetica, come quella richiesta per sintetizzare le catene oligosaccaridiche degli antigeni di gruppo sanguigno, sia del tutto inutile. Se così fosse, sarebbe difficile spiegare come mai le variazioni polimorfiche riguardino solo le estremità di queste catene, mentre tutto quello che riguarda le altre porzioni, la cui eventuale variabilità implicherebbe l'interruzione precoce delle catene oligosaccaridiche nascenti, è ben poco o addirittura niente affatto variabile. Questa constatazione si accorda solo con l'idea che la selezione è permissiva per variazioni che riguardano ‛la fogliolina', non la natura e l'esistenza dell'intero ramo.
Per quanto riguarda l'esistenza dei differenti gruppi sanguigni, a prima vista non sembra necessario considerare questo tipo di variabilità come un problema a sé rispetto a quello generale del significato biologico della variabilità genetica rilevata a livello molecolare (elettroforetica, di attività enzimatica, ecc.). Si potrebbe cioè supporre che il fatto che i polimorfismi degli antigeni eritrocitari siano stati scoperti con tecniche sierologiche, mentre per quelli enzimatici ci si è valsi dell'analisi elettroforetica, sia trascurabile quando si indaga sul loro significato biologico.
Tuttavia, mentre per molti polimorfismi elettroforetici e cromatografici ha senso chiedersi se la specie ‛si accorge' di queste variazioni, per i gruppi sanguigni la risposta è scontata in partenza: almeno il sistema immunitario percepisce questo tipo di differenze. Anzi, a guardar bene, ogni volta che si scopre un antigene si scopre che il sistema immunitario lo aveva scoperto prima di noi. Ciò non significa necessariamente che tutte le variazioni di gruppo siano funzionalmente rilevanti: le regioni e le proprietà strutturali di una molecola che sono più appariscenti per il sistema immunitario possono non essere quelle più importanti per la sua funzione. Nel caso di alcuni antigeni, però, il fatto stesso che il sistema immunitario sia in grado di accorgersi di loro è biologicamente rilevante, indipendentemente dall'eventuale effetto sulla funzione della molecola nel suo complesso. L'esempio più noto è il sistema Rh: anche se la funzione sconosciuta della molecola Rh è esplicata nello stesso modo da tutte le sue forme alleliche, questo polimorfismo è comunque biologicamente molto importante a causa della malattia emolitica Rh, che è appunto la conseguenza diretta del fatto che per il sistema immunitario non è indifferente se c'è o non c'è l'antigene D. E, sia pure in minor misura, lo stesso vale per il sistema AB0. Anzi, il ben noto effetto protettivo che l'incompatibilità materno-fetale per quest'ultimo sistema esercita su un feto Rh(+) di una donna Rh(−) si esplica anch'esso per via immunologica. Quindi il valore biologico di questi due sistemi e la loro interazione a livello popolazionistico sono dovuti al fatto che sono sistemi sierologici. Comunque è verosimile che situazioni in cui il significato biologico di un antigene di gruppo sanguigno coincide almeno in parte con il fatto di potersi comportare appunto come tale rispetto a individui della stessa specie siano l'eccezione più che la regola, dato che, almeno per quanto ne sappiamo, da esse non possono derivare che svantaggi, e ciò si concilierebbe molto male con la grande diffusione di questi polimorfismi. In particolare, l'esistenza del polimorfismo D-d, cioè Rh(+)-Rh(−), difficilmente può essere spiegata senza supporre che il rischio per i neonati Rh(+) figli di donne Rh(−) di andare incontro a una malattia emolitica Rh sia solo una parte della verità: se non ci fosse stato in gioco qualche altro fattore a noi sconosciuto, l'allele d - essendo meno comune del D - avrebbe dovuto scomparire, perché a ogni generazione questo meccanismo provoca la perdita di un pari numero di alleli D e d (ogni neonato morto a causa dell'incompatibilità con la madre, essendo necessariamente eterozigote Dd, corrisponde alla perdita di un allele D e di un allele d).
5. Il pattern di variabilità degli antigeni di gruppo sanguigno. - A. Gli antigeni proteici sono sintetizzati come tutte le altre proteine e quindi, almeno sotto questo aspetto, non vi è ragione di supporre che la loro variabilità presenti delle caratteristiche differenti da quella proteica in generale. Resta comunque fermo il fatto che la quota di variabilità che si svela come antigeni di gruppo sanguigno non è la stessa che si svelerebbe se le stesse proteine antigeniche venissero esaminate, ad esempio, con l'elettroforesi. Le capacità dei due metodi di svelare variazioni strutturali presentano delle limitazioni: limitazione regionale per la variabilità sierologica (solo la regione esterna delle sole proteine di membrana); limitazione chimica per la variabilità elettroforetica (solo le variazioni che comportano un cambiamento di carica elettrica, ma dell'intera molecola e di tutte le proteine). È possibile che in alcune proteine della membrana eritrocitaria la regione che sporge all'esterno abbia una funzione legata appunto a questa sua posizione (che costituisca, per esempio, un recettore specifico di membrana; che sia un antigene di differenziazione, ecc.). È, quindi, ragionevole supporre che il tipo di variazioni strutturali dimostrabili sierologicamente abbia un significato funzionale diverso da quello evidenziabile con altri metodi come quello elettroforetico.
B. Gli antigeni oligosaccaridici sono sintetizzati con processi che, oltre a essere molto più complicati di quelli della sintesi proteica, sono di natura tale da riflettersi sul loro pattern di variabilità, rendendolo molto diverso da quello delle proteine.
Presumibilmente, da questo punto di vista la differenza più importante tra i due meccanismi biosintetici è che mentre le unità di informazione degli mRNA, i codoni, specificano gli amminoacidi corrispondenti in modo del tutto indipendente dalla struttura primaria del tratto di catena polipeptidica nascente già sintetizzato, ogni unità di informazione di una catena biosintetica di un oligosaccaride, cioè ogni suo singolo enzima, per aggiungere il corrispondente monoso alla catena oligosaccaridica nascente deve riconoscere specificamente non solo quel monoso, ma anche il moncone a cui attaccarlo. Quindi, se in una certa posizione invece di un monoso ne viene attaccato un altro, è molto probabile che la crescita della catena oligosaccaridica si arresti a quel punto. In altri termini, è verosimile che tutte o quasi tutte le variazioni equivalenti a quelle che per un gene strutturale vengono chiamate missense oppure semplici delezioni intercalari risultino funzionalmente in terminazioni di catena. Naturalmente non si possono escludere - in contrasto con le mutazioni sense → term degli mRNA - delle compromissioni ‛parziali' dell'accrescimento ulteriore della catena oligosaccaridica, e questo dovrebbe verificarsi tutte le volte che il moncone di catena con il monoso ‛mutato' funzioni, sia pure con minore efficienza, da substrato accettore per l'enzima o per gli enzimi successivi.
È evidente allora che ci si deve aspettare un notevole grado di fissità evolutiva in tutta la sequenza dei vari tipi di catene oligosaccaridiche meno che nelle loro parti terminali; queste ultime, se cambiano, non implicano l'accorciamento della catena e non richiedono quindi una coevoluzione degli enzimi a valle dell'enzima mutato. Non si sa molto sulla lunghezza del tratto di moncone riconosciuto da questi enzimi glicosiltransferasici, cioè sul numero di enzimi che devono modificarsi per rendere evolutivamente accettabile una singola sostituzione carboidratica evitando l'interruzione dell'accrescimento sequenziale della catena oligosaccaridica. Se questo numero costituisse una frazione considerevole di una catena oligosaccaridica di lunghezza media (ordine di grandezza: 5-20 unità, comprese le ramificazioni) ne dovrebbe risultare un gradiente di variabilità sia intraspecie (grado di polimorfismo bilanciato e/o transeunte e/o di politipia) che interspecie (velocità di evoluzione filogenetica), si dovrebbe cioè passare dal massimo grado di variabilità genetica a livello delle estremità non riducenti delle catene oligosaccaridiche a un grado minore per il loro monoso preterminale (che richiede la coevoluzione del solo enzima terminale), a un grado ancora minore per il terz'ultimo monoso e cosi via. Non c'è dubbio che, effettivamente, quando la variabilità delle catene oligosaccaridiche di membrana è dovuta a polimorfismi genetici riguarda solo le loro parti terminali o preterminali (le proteine, invece, non presentano alcun pattern netto di distribuzione topografica della variabilità valido in generale, pur avendo ciascuna - naturalmente - un suo proprio pattern specifico). Inoltre, anche se il numero di sequenze oligosaccaridiche di glicoproteine e mucopolisaccaridi finora determinate non è molto grande - solo poche decine - non c'è dubbio che, se si confrontano vari tipi di oligosaccaridi suddivisi a seconda del tipo di legame (sempre β con cui si uniscono attraverso la loro estremità iniziale riducente alla catena polipeptidica, ne emergono solo due tipi di sequenze che nella loro estremità iniziale sono praticamente costanti anche per ciò che riguarda l'atomo di carbonio non 1 (non riducente) che vi è impegnato (sempre il 3 o sempre il 4).
La conservatività evolutiva di questi segmenti iniziali è sorprendente per l'ampiezza dei materiali biologici in cui la si osserva, sia dal punto di vista sistematico (microrganismi, piante, animali superiori) che da quello della differenziazione (glicoproteina antigelo di certi pesci dell'Antartide, immunoglobuline umane, sialoglicoproteine delle membrane eritrocitarie, fetuina, mucina gastrica, ecc.). Due ben noti fenomeni, l'esistenza degli anticorpi ‛naturali' e delle lectine, prima piuttosto misteriosi, trovano una semplice spiegazione alla luce di queste considerazioni.
Gli anticorpi ‛naturali' si trovano in individui che, almeno apparentemente, soddisfano solo il primo dei due requisiti specifici normalmente richiesti per produrre un anticorpo: il non possedere il corrispondente antigene e l'essere stati immunizzati verso di esso. Per i soggetti A, B e 0 il non possedere gli antigeni B, A, A e B, rispettivamente, sembra essere condizione necessaria e anche sufficiente per produrre i corrispondenti anticorpi. È stato ormai dimostrato che il meccanismo di produzione di queste agglutinine è, contrariamente alle apparenze, quello comune a tutti gli altri anticorpi: gli antigeni AB0 sono ubiquitari, e in particolare si trovano sulle pareti batteriche di vari ceppi di Escherichia coli, il microrganismo più abbondante del nostro intestino crasso; occasionalmente esso passa in circolo (batteriemia asintomatica), per cui tutti sono esposti al contatto con questi antigeni anche se, naturalmente, solo coloro che non li possiedono producono, per effetto di questa esposizione, gli anticorpi corrispondenti. Si tratta quindi di un fenomeno relativamente banale (certo non dal punto di vista clinico, comunque). Ma a guardar bene ci si accorge che è banale solo il meccanismo di produzione di questi anticorpi ‛naturali', non l'universalità della diffusione degli antigeni che li hanno indotti: perché certi antigeni come quelli del sistema AB0, Hh e P sono tanto diffusi da causare la regolare comparsa di anticorpi naturali, mentre altri come quelli del sistema Rh ed MNS hanno una diffusione filogeneticamente tanto più limitata? Se si tiene conto che i determinanti antigenici dei primi sono di natura oligosaccaridica mentre quelli dei secondi sono almeno in parte di natura proteica (per il sistema MNS forse gioca un ruolo anche la componente oligosaccaridica), sembra ragionevole supporre che ciò dipenda dalla fissità filogenetica dei primi in contrapposizione alla plasticità dei secondi.
Le lectine sono proteine vegetali nella cui molecola si trovano una o più regioni aventi una conformazione stericamente complementare a quella di certe strutture carboidratiche della superficie dei globuli rossi; esse sono quindi capaci di legarsi a tali strutture in modo specifico, eventualmente funzionando da ponte tra strutture uguali poste su globuli rossi diversi, proprio come le agglutinine eritrocitarie. Se la struttura chimica riconosciuta da una di queste sostanze è comune ai globuli rossi di tutti gli individui, la lectina funziona come un'agglutinina universale (il più tipico esempio è la ben nota fitoemoagglutinina o concanavalina, il cui recettore si trova anche sui linfociti, sui quali esercita un potente effetto mitogeno); se invece è il prodotto di un allele di gruppo sanguigno, la lectina funziona come se fosse un'agglutinina specifica (lectine anti-H, anti-A, anti-A1, anti-M, anti-N). Una così perfetta e specifica complementarietà tra queste proteine vegetali e questi determinanti suggerisce che questi ultimi non siano del tutto estranei per le piante che producono le lectine: una nuova indicazione della loro distribuzione universale. E anche questi sono determinanti oligosaccaridici e non proteici (o, almeno, hanno una componente oligosaccaridica come gli antigeni M ed N della glicoforina).
Una diretta implicazione di queste considerazioni è che la scoperta di anticorpi naturali e di lectine specifici verso un determinato antigene deve essere considerata come una indicazione che esso è almeno in parte di natura oligosaccaridica e, viceversa, la dimostrazione dell'assenza di tali anticorpi naturali e il non aver trovato nessuna lectina specifica per un certo antigene suggeriscono che esso sia di natura proteica.
3. Genetica del siero.
a) Generalità.
Il siero è ciò che rimane del plasma dopo che si è verificata la coagulazione. La genetica del siero e quella del plasma coincidono in pratica con la genetica delle sue proteine, quasi tutte (sul piano quantitativo ponderale, non come numero di tipi) sintetizzate nel fegato, con l'importantissima eccezione delle immunoglobuline.
La gamma delle concentrazioni dei vari tipi di proteine plasmatiche è amplissima: si va dai valori addirittura infinitesimali di alcuni ormoni proteici fino alla concentrazione circa millesimomolare della sieroalbumina (circa 7 g per 100 rnl di siero). Le concentrazioni di alcune delle più note proteine plasmatiche sono presentate nella fig. 15.
La genetica delle sieroproteine può essere per la massima parte perfettamente inquadrata, per i suoi aspetti molecolari, nel modello ben più completo del sistema dell'Hb, e per quelli formali e popolazionistici nella genetica dei polimorfismi enzimatici eritrocitari. Rientrano in questo gruppo soprattutto le aptoglobine e l'α1-antitripsina, polimorfismi genetici interessanti, specialmente il secondo, di cui si conoscono due alleli comuni ipofunzionanti a cui è associata una particolare predisposizione all'enfisema polmonare. Di tutti questi polimorfismi non si tratterà, dato che al momento attuale non presentano problemi specifici di rilevanza biologica generale.
Fanno invece storia a sé, oltre alle immunoglobuline, anche le proteine del complemento e della coagulazione. Questi ultimi due sistemi, pur essendo adibiti a funzioni completamente diverse (il primo è l'effettore chimico di molte reazioni immunologiche, il secondo è uno dei principali responsabili dell'emostasi, cioè dell'insieme dei processi incaricati di arrestare o comunque limitare la fuoriuscita del sangue da vasi sanguigni lesi) sono tuttavia molto simili per alcuni aspetti. Uno dei più importanti è la loro estrema complessità sia strutturale (numero di fattori coinvolti) sia funzionale, a prima vista - ma solo a prima vista - gratuita. In entrambi il processo di innesco viene seguito da una vera e propria cascata di reazioni (donde il termine di ‛reazioni a cascata') ognuna delle quali è causa della successiva, ma spesso influenza l'andamento di molte delle precedenti e di alcune delle non immediatamente successive. Si viene così a creare una rete inestricabile di interazioni complesse e di reazioni controllate a monte e a valle. La ragione di tanta complessità invece della semplice trasformazione del fibrinogeno in fibrina (per la coagulazione) o della lisi dei globuli rossi o dei Batteri (per il complemento) è dello stesso tipo di quella che ha consigliato di non adottare come sistema di frenaggio delle automobili una zeppa che, incastrandosi tra le ruote, le possa bloccare di colpo. La frenata sarebbe efficace, ma i passeggeri della macchina non vivrebbero certo a lungo. Processi di questo genere devono poter essere estremamente tempestivi ed efficaci, ma è altrettanto necessario che siano tenuti sempre sotto controllo, che siano modulabili, accelerabili e frenabili. La necessità di disporre di reazioni esplosive controllate rende conto del significato biologico della loro complessità (ad es., come potrebbe essere arrestato un processo di coagulazione che consistesse semplicemente in una trasformazione catalitica del fibrinogeno in fibrina?) e della loro altrettanto complicata genetica, solo in parte delucidata.
b) Le immunoglobuline (Ig), o anticorpi circolanti.
1. Il sistema immunitario: un sistema unico per versatilità e grado di amplificazione differenziale di singole funzioni. Le Ig sono glicoproteine con attività di anticorpi (v. immunologia e immunopatologia: Immunologia generale). È noto da molto tempo che la presenza di qualsiasi sostanza estranea e complessa nell'ambiente interno di un vertebrato è seguita dopo un certo periodo (dell'ordine di una decina di giorni) dalla comparsa, nel siero di questo organismo, di sostanze di natura proteica dette anticorpi (Ab, dall'inglese Antigen-binding), capaci di combinarsi specificamente con questa sostanza estranea riconosciuta come non-self, e che la comparsa degli anticorpi in circolo è a sua volta seguita dalla scomparsa del non-self. Quest'ultimo, per il fatto di aver provocato la produzione dell'anticorpo corrispondente, è chiamato antigene (Ag, dall'inglese Antibody-generating).
Dopo un certo tempo dalla scomparsa dell'antigene, scompare anche l'anticorpo. Qualcosa tuttavia è rimasto, il ricordo specifico della passata esperienza immunologica (memoria immunitaria): se lo stesso organismo, ormai privo di anticorpi circolanti diretti contro l'antigene a cui era stato esposto, è nuovamente trattato con lo stesso antigene anche a dosi molto piccole (il cosiddetto richiamo), si assiste a una risposta immunitaria molto più rapida, specifica ed efficace; infatti, dopo soli due-tre giorni compaiono di nuovo gli anticorpi verso quell'antigene nei cui confronti mostrano un'affinità chimica particolarmente elevata, e in breve tempo arrivano a titoli molto più alti di quelli raggiunti con la prima immunizzazione.
Due caratteristiche distinguono in maniera nettissima il sistema immunitario da qualsiasi altro: a) la sua versatilità, cioè l'ampiezza, reale e potenziale, dello spettro delle molecole qualitativamente diverse che esso produce o potrebbe produrre, cioè i diversi anticorpi; b) i fattori di amplificazione che possono entrare in gioco, cioè di quanto può essere incrementata la sintesi di uno specifico anticorpo. Infatti, il numero di anticorpi che possono essere o non essere prodotti è almeno dell'ordine delle migliaia, ma probabilmente molto di più, e i fattori moltiplicativi che possono essere implicati nella sintesi preferenziale di uno qualsiasi di questi innumerevoli tipi di Ig sono dell'ordine dei miliardi. Inoltre, poiché la sintesi preferenziale di un anticorpo è sostenuta, come vedremo, non solo da un'elevata attività sintetica delle cellule capaci di produrre quel particolare anticorpo, ma anche dalla loro attiva moltiplicazione, questo estremo grado di capacità potenziale di amplificare una certa funzione fra le tante possibili comporta anche la necessità che i meccanismi di controllo specifico della moltiplicazione cellulare operino in questo sistema su ambiti di parecchi ordini di grandezza: i cloni stimolati, pur restando sotto controllo (per esempio, in funzione della concentrazione delle Ig circolanti che essi hanno prodotto), devono poter tenere un ritmo moltiplicativo quale si riscontra solo in alcuni tumori e nei tessuti embrionali, mentre gli altri doni immunocompetenti - simili a quello in tumultuosa moltiplicazione, salvo che per la specificità dell'anticorpo che sono capaci di produrre - si moltiplicano molto poco o niente affatto.
Il significato biologico di queste due proprietà esclusive del sistema immunitario è facilmente individuabile: esse rappresentano l'unica risposta possibile alla imprevedibilità praticamente totale di quella che potrebbe essere la natura chimica di un non-self. Infatti l'essere in grado di produrre una risposta specifica verso qualsiasi materiale estraneo possibile richiede obbligatoriamente una versatilità di grado estremo; d'altra parte, affinché le poche risposte specifiche che effettivamente dovranno essere espresse risultino efficaci, è necessario che siano amplificabili senza limiti, cioè fino a che l'anticorpo prodotto non abbia neutralizzato tutto l'antigene circolante, che è come dire fintantoché non ci sarà anticorpo libero in circolo (che infatti in certe condizioni inibisce l'ulteriore crescita e attività secretoria del clone che lo ha prodotto).
2. Il problema immunologico ai livelli proteico (la base molecolare della duplice natura degli anticorpi circolanti) e cellulare. A. Il problema a livello proteico: gli anticorpi come ‛enzimi mancati'. Le Ig costituiscono una categoria di proteine allo stesso tempo eterogenea e omogenea.
Eterogenea, anzi estremamente eterogenea, perché la loro capacità complessiva di combinarsi, in modo altamente specifico e con elevata affinità, a un numero grandissimo di antigeni è dovuta al fatto che in ogni individuo esse sono costituite da una miscela di vari tipi di molecole, ciascuno dei quali riconosce specificamente un determinato antigene. In altre parole, la polispecificità anticorpale di ogni siero è l'espressione del gran numero di tipi di molecole monospecifiche in esso contenute.
Omogenea, perché quello che segue all'evento specifico della combinazione di un dato anticorpo al corrispondente antigene è invece molto aspecifico: di regola consiste nella distruzione del complesso Ab-Ag da parte di effettori aspecifici (che possono essere tanto cellule, ad es. macrofagi, che insiemi di molecole, ad es. il sistema del complemento), e quindi, in ultima analisi, del materiale non-self giunto nell'ambiente interno dell'organismo. E, dato che questi effettori aspecifici sono in grado di riconoscere e distruggere qualsiasi complesso Ab-Ag, indipendentemente dal tipo di antigene in esso contenuto, si deve postulare che la struttura comune che essi riconoscono nei complessi Ab-Ag sia una parte della molecola anticorpale, e questa deve pertanto avere una struttura simile in tutti gli anticorpi - quale che sia la loro specificità - che si siano legati all'antigene corrispondente (v. fig. 16).
Ogni anticorpo può quindi essere considerato, nei riguardi dell'antigene corrispondente, come un ‛enzima mancato': si combina con esso altrettanto specificamente di un enzima con il suo substrato, ma senza modificarlo, cioè senza distruggerlo. Di ciò è incaricato il sistema effettore aspecifico. Solo i due componenti insieme, l'enzima mancato e l'effettore ‛aspecifico' (in quanto capace di collaborare con tutti gli enzimi mancati), in definitiva sono in grado di portare a termine il loro compito, la distruzione del non-self. Ma l'evoluzione non avrebbe potuto realizzare un sistema costituito da un numero di enzimi ‛completi' così grande da poter riconoscere, e anche degradare, qualsiasi substrato. Se per semplicità limitiamo questo discorso agli antigeni di natura proteica, ci si accorge subito che le proteasi veramente specifiche (che, cioè, catalizzano una particolare reazione, l'idrolisi di un legame peptidico, e solo in un determinato punto di una determinata molecola) sono l'eccezione più che la regola: esse fanno parte di sistemi altamente specializzati, come quelli del complemento e della coagulazione. Al di fuori di questi ci sono due tipi di molecole almeno potenzialmente capaci, da sole o in collaborazione con altre sostanze, di distruggere le proteine: le proteasi aspecifiche (come la pepsina, la tripsina, le catepsine ecc.) e, appunto, gli anticorpi contro antigeni proteici. Entrambi vanno tenuti sotto controllo: le proteasi aspecifiche non hanno modo di agire che in certi compartimenti programmati (il lume intestinale o l'interno dei lisosomi); gli anticorpi, che invece devono per forza avere accesso all'ambiente interno dell'organismo, visto che il loro compito è proprio quello di scoprire e causare la distruzione del non-self che vi sia eventualmente capitato, sono controllati a monte, cioè prevenendo, attraverso un meccanismo complesso indicato con il termine di ‛tolleranza immunitaria', la produzione degli anticorpi contro il self; in questo modo viene conferita al sistema immunitario nel suo insieme una sola capacità discriminativa, quella tra self e non-self, che è l'unica biologicamente rilevante.
La base molecolare dell'omogeneità della funzione è stata trovata con la scoperta che tutte le Ig hanno in comune una struttura di base, il cosiddetto ‛monomero immunoglobulinico' H2L2, costituito da due catene polipeptidiche pesanti (H, heavy) e da due leggere (L, light). Alcune Ig sono monomeriche, altre dimeriche e altre pentameriche (v. tab. V e fig. 16; v. immunologia e immunopatologia: Immunologia generale).

La tab. V mostra il grado di eterogeneità riconoscibile in tutti i sieri umani, che è del tutto indipendente dall'ampio spettro delle sue specificità anticorpali. Come si vede, esso è relativamente modesto: poco più di una decina di classi o sottoclassi di catene pesanti, a cui corrispondono altrettante classi o sottoclassi Ig, ognuna con una sua propria catena H, e tre tipi o sottotipi di catene L, che sono invece comuni a tutte le Ig.
In conclusione, solo una ventina di anni fa molto era stato chiarito sulla base strutturale di quanto c'è di comune in tutte le Ig, mentre non si sapeva ancora nulla della natura chimica della loro eterogeneità come anticorpi, cioè come molecole capaci di riconoscere ciascuna un antigene specifico, salvo che anche per questa una base strutturale doveva pur esserci. Né si prevedeva allora di arrivare a chiarirla in pochissimi anni - come in effetti è accaduto - perché il problema era di quelli che, se non ci si imbatte in una situazione imprevista molto favorevole, appaiono intrinsecamente irrisolvibili. La sua stessa natura lo rendeva tecnicamente non affrontabile: si può determinare la struttura primaria, cioè la sequenza amminoacidica di una proteina pura, non quella di un miscuglio estremamente eterogeneo di proteine, quale doveva essere l'insieme delle Ig. Questa volta la circostanza imprevista che ha permesso di determinare le sequenze amminoacidiche di svariate Ig pure non è stata la messa a punto di una nuova tecnica, bensì la scoperta che in certe condizioni (cioè nei mielomi) queste sono facilmente disponibili in grandi quantità.
B. La soluzione del problema: le catene H e le catene L consistono di due regioni, una variabile e una costante. Si sapeva da molto tempo che il siero degli individui affetti da mieloma, un tumore delle plasmacellule produttrici di Ig, contiene una grande quantità di Ig e che in ogni malato questo eccesso di Ig è da attribuirsi a una frazione immunoglobulinica considerevolmente omogenea: non si tratta cioè di un generico eccesso di Ig, bensì di un eccesso di un particolare tipo di Ig. Questi malati, inoltre, eliminano con le urine addirittura grammi di una proteina, detta proteina di Bence-Jones, che non è altro che un dimero di catene L, e anche questa appariva omogenea, a differenza dei dimeri di catene L che si potevano ottenere partendo da un siero umano qualsiasi.
L'intuizione fondamentale di Edelman (premio Nobel 1972) è stata che ogni mieloma è il risultato della crescita tumorale di un solo clone di cellule e che il motivo della omogeneità chimica della frazione Ig in eccesso è che questa è il prodotto di quel clone tumorale. Si avevano già a quel tempo ottime ragioni sia teoriche sia sperimentali per pensare che ogni plasmacellula producesse molecole Ig tutte con la stessa specificità anticorpale. Bastava a questo punto estrapolare questa idea dalle cellule ai cloni per giungere all'ipotesi che in ogni individuo portatore di mieloma la grandissima quantità della frazione Ig in eccesso (facilmente separabile dalle restanti Ig del suo siero) e della proteina di Bence-Jones (già allo stato puro nelle sue origini) erano proteine omogenee. È stato facile dimostrare che questa ipotesi era esatta. Questo ha significato che da quel momento ogni individuo portatore di mieloma ha costituito la fonte di una particolare Ig praticamente allo stato puro e in quantità tali che se ne è potuta determinare la struttura primaria: diventava finalmente possibile confrontare fra loro Ig varie.
Il quadro che ne è emerso (v. fig. 16) rende conto perfettamente delle proprietà funzionali degli anticorpi: in ogni monomero H2L2 tanto la catena H che la L consistono di due regioni, una iniziale di circa 110 residui amminoacidici (a partire dal residuo NH2-terminale), variabile, e una, la successiva, di circa 110 residui nelle catene L e di circa 330 (≃3×110) nelle Hγ, costante. Cioè, il tratto iniziale delle catene L e delle H differisce da una Ig a un'altra, mentre il tratto successivo è uguale per tutte le Ig che appartengono alla stessa classe, sottoclasse, tipo e allotipo. Diventava allora possibile definire in termini molecolari, e non solo sierologico-funzionali, i tre tipi di eterogeneità che si conoscevano per le Ig: a) isotipia, l'insieme delle differenze esistenti tra classi e sottoclassi diverse di Ig, riconoscibili o a livello antigenico (cioè con antisieri specifici anticlasse e sottoclasse) o funzionale (capacità di legarsi al complemento, di traversare la placenta, ecc.); tutti gli individui normali della stessa specie hanno Ig di tutte le classi e sottoclassi e le regioni costanti delle Ig della stessa classe e sottoclasse sono tutte eguali anche se appartengono a individui diversi (salvo eventuali differenze allotipiche) o a molecole con specificità diversa come anticorpi; b) allotipia, l'insieme delle differenze tra parti costanti di Ig della stessa classe e sottoclasse, dovute all'esistenza di alleli dei geni strutturali corrispondenti; c) idiotipia, l'insieme delle differenze tra le parti variabili delle Ig, espressione dell'eterogeneità delle Ig come anticorpi.
Si era inoltre in grado di precisare i livelli di questi diversi tipi di eterogeneità e di comprenderne il significato. Non esiste alcuna eterogeneità tra le due semimolecole HL che costituiscono un monomero H2L2. Questa uguaglianza - che è un requisito indispensabile per il funzionamento di qualsiasi molecola anticorpale (essa funziona facendo da ponte tra siti antigenici uguali) - è spiegata dal fatto che ogni plasmacellula, anzi ogni clone di plasmacellule, produce catene H con un unico idiotipo e catene L anch'esse con un unico idiotipo, e quindi molecole con un'unica specificità anticorpale. È vero che nel corso della maturazione di ogni clone si assiste di regola a un switch da una classe a un'altra (all'inizio i doni producono IgM [o JgD], e in seguito IgG [o IgE]), ma questo riguarda solo le regioni costanti delle molecole Ig che esso produce e non le regioni variabili, sia H sia L. Inoltre, studiando eterozigoti per regioni costanti appropriate, si è accertato che si verifica anche il fenomeno della cosiddetta ‛esclusione allelica', cioè ogni clone esprime uno solo dei due alleli del gene che ha deciso di far funzionare. I vari doni di plasmacellule committed, cioè ‛impegnate', non sono quindi unità funzionali equivalenti e intercambiabili come, per esempio, i nefroni e gli infundiboli polmonari, perché tra di loro intercorrono differenze qualitative. Il sistema immunitario è un esercito di specialisti, ciascuno dei quali (i cloni ormai differenziati) è irreversibilmente capace di combattere un solo tipo di nemico, un particolare antigene, perché è in grado di vedere solo quello e di costruire e secernere - dopo essersi accorto della sua presenza - un solo tipo di arma, l'anticorpo specifico per quell'antigene. La versatilità immunologica di un organismo - difficile da misurare, ma comunque certamente grandissima - è dovuta quindi al grandissimo numero di doni monospecifici, diversi l'uno dall'altro, che vi si trovano. È una proprietà dell'insieme, non dei suoi componenti.
In ogni singolo organismo, invece, esistono almeno due tipi di eterogeneità: quella idiotipica, cioè di specificità anticorpale, tra le Ig prodotte da doni diversi, e quella isotipica, cioè tra classi e sottoclassi. In molti individui, inoltre, cioè negli eterozigoti per le regioni di DNA che specificano una o piu' regioni costanti, ne può esistere anche una terza, quella allotipica.
È evidente che l'eterogeneità tra Ig di soggetti diversi è ancora maggiore, non solo perché le loro ‛storie immunologiche' sono certamente diverse, ma anche perché spesso sono diversi i genotipi per i geni polimorfici delle parti costanti. E questo, naturalmente, vale ancora di più se si tratta di individui di razze diverse, come per qualsiasi altro polimorfismo genetico. Anzi, con l'eccezione dell'HLA, l'insieme dei polimorfismi per le Ig costituisce il gruppo di marcatori genetici e antropologici più variabili e quindi più discriminanti che si conoscano.
C. Anatomia e fisiologia della molecola immunoglobulinica. Alla conoscenza delle sequenze amminoacidiche di svariate catene L e di alcune catene H è seguita anche quella dell'architettura sterica tridimensionale della molecola Ig nativa e funzionante; è stato così possibile identificare le regioni o ‛domini' responsabili delle svariate funzioni che fino a quel momento erano state attribuite globalmente all'intera molecola. Si è riusciti cioè, anche se non in misura così completa come per l'Hb, a costruirne l'‛anatomia funzionale' a livello dei vari organi submolecolari (v. fig. 16). È risultato in tal modo evidente che, come si era potuto ipotizzare dall'analisi delle sequenze amminoacidiche, ogni molecola Ig è suddivisa in domini ben individuabili a livello tridimensionale: ogni dominio ha una conformazione globulare compatta ed è connesso a quello adiacente, o ai due adiacenti, da un tratto di molecola non rigido, cioè privo di struttura secondaria. Il primo dominio della catena L, quello variabile, forma insieme al dominio variabile della catena H un sito anticorpale o sito Ab. Ogni monomero è costituito da due metà identiche tenute insieme soprattutto da ponti disolfuro che giocano un ruolo importante anche per quella che può essere chiamata l'architettura ‛intradominio'. Nell'insieme un monomero ha una forma simile a una Y, con una regione fondamentale situata all'origine dei rami della Y, la cosiddetta ‛cerniera' (hinge); questa non solo è la sede dei legami S-S che tengono unite le due catene H e quindi le due metà della molecola, ma soprattutto è dotata, a causa dell'abbondanza di residui di prolina, di una particolare flessibilità. In questo modo alle due antigen-binding arms è concesso un gioco di mobilità angolare relativa, sufficiente a conferire a ciascun braccio una certa autonomia nel cercare l'adattamento sterico migliore con il sito antigenico corrispondente. Questo ‛accorgimento submolecolare' aumenta di molto la proporzione di molecole anticorpali funzionalmente efficaci, cioè in grado di ancorarsi a molecole antigeniche diverse formando così complessi Ab-Ag di alto peso molecolare, che sono i veri substrati degli organi effettori aspecifici.
3. Il problema immunologico a livello genetico dopo aver dovuto scartare le teorie ‛istruttive'. La delucidazione della struttura delle Ig attraverso lo studio dei mielomi aveva risolto il problema della duplice natura degli anticorpi: parte effettrice aspecifica, identificabile nella sezione costante delle catene H; parte specifica, identificabile nelle sezioni variabili H ed L. Inoltre, la scoperta che ogni cellula produce un unico tipo di sezione variabile L e un unico tipo di sezione variabile H aveva spiegato come mai le due o più valenze di ogni molecola anticorpale sono identiche, per cui riconoscono gli stessi determinanti antigenici. Queste due caratteristiche irrinunciabili degli anticorpi erano state quindi spiegate a livello sia proteico, cioè di struttura delle molecole Ig, sia delle cellule e dei doni produttori di queste molecole.
Ma la soluzione di questi problemi ne prospettava a monte uno ancora più complesso che riguardava la base genetica di queste proprietà proteiche e cellulari indispensabili per il corretto funzionamento del sistema immunitario. Più precisamente, si trattava di capire perché cellule di cloni diversi producano Ig che nelle regioni variabili sono differenti da quelle degli altri cloni, mentre nelle regioni costanti sono uguali a quelle di tutti gli altri - numerosissimi - cloni dello stesso tipo (per le catene L), classe e sottoclasse (per le catene H).
È evidente che non ci sarebbero state difficoltà se si fosse stati disposti ad accettare una qualsiasi teoria ‛istruttiva', cioè che le cellule destinate a produrre gli anticorpi potessero ricevere l'istruzione, o meglio l'informazione, di come produrre un anticorpo verso un determinato antigene dall'antigene stesso. Ma questa ipotesi era stata scartata come ‛impossibile' dal punto di vista molecolare: come avrebbe potuto un determinante antigenico causare la produzione di una sequenza di DNA specificante una sequenza amminoacidica che, dopo aver raggiunto la sua conformazione sterica tridimensionale definitiva, fosse a esso complementare? Si era così giunti alla convinzione che, qualsiasi fosse il meccanismo responsabile della capacità del sistema immunitario di rispondere adeguatamente a qualunque antigene, il ruolo di quest'ultimo poteva essere solo ‛selettivo': in base a questa teoria, ogni antigene provoca la produzione dell'anticorpo corrispondente stimolando selettivamente il clone o i cloni monospecifici preesistenti che costituiscono la base cellulare dell'ampiezza del repertorio del sistema immunitario. Ma i meccanismi responsabili dell'esistenza di questo repertorio non hanno nulla a che vedere con gli antigeni.
Abbandonate le teorie ‛impossibili', si è passati quindi alle ‛quasi impossibili': non appena si è tentato di speculare sui meccanismi che potrebbero rendere il sistema immunitario capace di fare quello che indubitabilmente fa, è diventato necessario rimettere in discussione caratteristiche della struttura e del modo di funzionare del genoma che si consideravano ormai definitivamente acquisite. È stato cioè inevitabile porsi quesiti che, per la genetica classica di quell'epoca tutt'altro che lontana (solo una quindicina di anni fa), potevano a ragione essere definiti ‛eretici'. I ‛dogmi' da mettere in discussione erano due, e ad almeno uno si doveva rinunciare.
A. Il primo quesito ‛eretico': cloni diversi esprimono geni diversi perché hanno fatto una scelta diversa nell'ambito di genomi uguali (ipotesi ‛ortodossa') oppure perché contengono geni diversi (ipotesi ‛eretica')? L'eresia di quest'ipotesi consiste nel considerare possibile che un tipo di differenziazione si verifichi a livello della struttura, e non semplicemente dell'espressione, del DNA.
Di regola, nel caso di cellule differenziate di uno stesso organismo, cioè originate tutte dallo stesso zigote da cui hanno derivato il proprio patrimonio genetico solo per mitosi, si dà per scontato che possiedano gli stessi geni. Ma in questo caso non si poteva fare a meno di dubitarne. Infatti, ogni organismo può generare anticorpi verso qualsiasi antigene, anche verso quelli mai esistiti in natura o che non hanno mai avuto l'opportunità di immunizzare alcun organismo di nessuna specie; quindi, il prezzo richiesto per restare fedeli al dogma che in ‛tutte' le cellule di un organismo deve esistere almeno un gene strutturale per ogni catena polipeptidica che viene sintetizzata è molto alto: occorre accettare che l'evoluzione abbia permesso il mantenimento in tutte le specie dei Vertebrati (e quindi per tempi estremamente lunghi) di un repertorio grandissimo di geni strutturali, tanti che costituirebbero nel loro insieme una parte cospicua dell'intero genoma, che sarebbero stati quasi tutti sempre inutili.
A guardar bene, si tratta, in realtà, non di un solo dogma, ma di due dogmi. Il primo dice che una cellula, se produce una certa catena polipeptidica, contiene una sequenza di DNA che specifica la sequenza amminoacidica di quel polipeptide. E di questo non è il caso di dubitare: il sistema immunitario è sì capace di prestazioni eccezionali, ma non è ‛magico'. Il secondo afferma che tutte le cellule somatiche di uno stesso organismo hanno gli stessi geni, per cui se anche un solo clone di cellule somatiche produce una certa catena polipeptidica, vuol dire che tutte le cellule dell'organismo - cioè anche quelle che non la producono - contengono il gene strutturale per quella catena polipeptidica.
Ebbene, per evitare l'assurdo evoluzionistico che si è appena illustrato, bastava rinunciare solo al secondo di questi dogmi, limitandosi ad affermare che i geni strutturali per una certa Ig sono posseduti solo dalle cellule che producono quella Ig. In altre parole, esisterebbe una variabilità genetica - e non semplicemente di espressione differenziale di genomi identici - tra i diversi cloni immunocompetenti di uno stesso organismo e questa variabilità genetica sarebbe di necessità postzigotica: l'evoluzione non avrebbe affatto mantenuto indefinitamente un numero grandissimo di geni inutili come patrimonio comune di tutti i gameti e di tutte le cellule somatiche di tutti gli organismi di tutte le specie dei Vertebrati, bensì solo la potenzialità di generarne uno per ciascuno dei numerosissimi doni che si formano nel corso della differenziazione del sistema immunitario. Ogni organismo conterrebbe sì moltissimi geni - di qui la sua grandissima versatilità immunologica - ma ciascuna delle sue cellule ne avrebbe uno solo (o, comunque, pochi).
B. Il secondo quesito ‛eretico': per specificare la sequenza amminoacidica di ogni singola catena H ed L sintetizzata da una cellula immunocompetente c'è nei gameti e nello zigote un solo gene strutturale (ipotesi ‛ortodossa') oppure ve ne sono più di uno (ipotesi ‛eretica')?
L'esistenza di polimorfismi genetici per molte delle catene costanti L ed H, ciascuno dei quali si comporta come un polimorfismo per un singolo gene, dimostra senza alcun dubbio che ogni particolare tipo di catena costante è codificato da un singolo gene strutturale (un gene per gamete e due per cellula diploide). Ciò non sarebbe inconciliabile con l'ipotesi che alla base della variabilità tra gli anticorpi prodotti dai differenti cloni vi sia una variabilità di origine postzigotica tra i loro genomi: basta infatti postulare che ognuno dei pochissimi geni destinati alla sintesi delle Ig (uno per tipo di catene L e per classe e sottoclasse di catene H) sia costituito da una sezione con altissima mutabilità somatica (la sezione V) e da una sezione (la sezione C) che si comporti invece come tutti gli altri geni, rimanendo cioè immodificata nel corso delle generazioni cellulari somatiche.
L'altra ipotesi - quella che, per rimanere fedele al dogma che i fenomeni differenziativi di un organismo si svolgono solo a livello di espressione differenziale di informazioni preesistenti nel DNA di tutte le sue cellule, esclude una variabilità postzigotica - deve invece rinunciare a un altro dogma, quello che afferma che ogni singola catena polipeptidica è codificata da un gene strutturale che è singolo non solo nella cellula che produce quella catena polipeptidica, ma anche nei gameti e quindi nello zigote e in tutte le cellule dell'organismo. Infatti la fedeltà al primo dogma obbliga a postulare l'esistenza nei gameti di un numero grandissimo di sequenze di DNA per le parti variabili (malgrado l'inutilità evolutiva della grande maggioranza di esse), ma poiché resta comunque il fatto che i geni per le parti costanti sono pochissimi, diventa inevitabile accettare l'idea che i geni strutturali che operano la sintesi delle catene Ig nelle cellule immunocompetenti si sono originati dalla fusione di una delle numerosissime sequenze per una parte variabile con una delle pochissime per una parte costante, generandosi in questo modo un gene completo VnCn prima inesistente e diverso per la sezione V dai geni completi VC degli altri doni (donde la loro grandissima eterogeneità idiotipica).
In conclusione non si poteva fare a meno di accettare almeno una delle due ‛eresie': o che il gene per una singola catena polipeptidica è uno, ma l'informazione a livello di DNA per una parte di essa non è trasmessa con i gameti bensì prodotta in seguito in un clone somatico; oppure che questa informazione è già presente nei gameti, ma che per produrre una singola catena polipeptidica Ig ci siano nei gameti due geni che si fonderebbero, generando quello definitivo unico, solo in seguito nei capostipiti di tutti i cloni produttori di Ig.
4. La soluzione del problema immunogenetico. Cloni diversi producono Ig con parti variabili H ed L diverse perché possiedono geni VL e VH diversi, generatisi dalla fusione - che può verificarsi in modi diversi - di uno dei tanti geni V con uno dei geni J (e, nel caso delle VH, con uno dei geni D). Ciascuno di questi geni V diversi, che è posseduto in esclusiva da un clone immunocompetente, si fonde infine con un gene C dando origine in tal modo a un gene VC completo e funzionante.
Questa è la risposta, ormai certa, ai quesiti immunogenetici.
Tutte le ‛eresie' ipotizzate, anche le più azzardate, erano quindi meno estreme della realtà, che è risultata un miscuglio delle due ipotesi appena illustrate: non solo la variabilità idiotipica interclonale è dovuta al fatto che cloni diversi hanno geni diversi per le parti variabili delle loro Ig, ma ciascuno di questi geni diversi è prodotto dalla fusione di geni differenti, fusione che può inoltre verificarsi in vari modi (anche a parità di geni che si saldano assieme), generandosi così un'ulteriore variabilità genetica postzigotica.
Anche se non si può dire che proprio tutto è stato chiarito sulla struttura e sul modo di funzionare delle regioni genetiche responsabili della produzione delle Ig, il quadro è ormai chiaro nelle sue linee generali e le ipotesi hanno ceduto il passo a fatti accertati; si tratta per la massima parte di progressi recentissimi, conseguiti all'impiego di nuove tecniche che hanno finalmente permesso di studiare il DNA direttamente, invece che solo per inferenza da un fenotipo da esso prodotto, cioè dalla struttura delle Ig.
A. La struttura delle regioni genetiche deputate alla sintesi delle catene H ed L delle Ig, all'origine (cioè nello zigote). L'analisi di queste strutture ha potuto mettere in evidenza alcune peculiarità molto importanti, quali il considerevole numero di geni capaci di codificare per la maggior parte delle regioni variabili; un'altra peculiarità di queste regioni - del tutto inattesa - è che per codificare la parte variabile delle catene Ig non esiste ‛semplicemente' una serie di geni V, ma anche una serie, sia pure molto limitata, di altri geni, i geni J (per Junction tra V e C), e, nel caso delle VH, anche un'altra serie di geni, i geni D (per Diversity). Insomma, il gene strutturale incaricato della sintesi di una catena H o di una catena L non deriva ‛solo' da due geni, quello per la sua regione V e quello per la sua regione C (per la quale, come aveva dimostrato la genetica formale, ne esiste effettivamente uno solo per famiglia isotipica di Ig), ma addirittura da tre o quattro geni distinti, che a un certo punto si trovano tutti a cooperare alla sintesi di una singola catena polipeptidica.
Un'altra proprietà molto interessante è l'ordine in cui sono disposti i geni CH. Come per i clusters emoglobinici, questi geni si succedono nello stesso senso della trascrizione e secondo la sequenza con la quale sono destinati a funzionare nel corso dello sviluppo differenziativo di ogni singolo clone; tale successione è anche la stessa con la quale questi geni cominciano a funzionare nel corso dello sviluppo dell'intero organismo e, nel suo complesso, coincide con l'ordine di comparsa di questi geni nella scala dei Vertebrati. Infatti, i vertebrati inferiori producono solo IgM, che possono a ragione essere considerate forme primitive di immunoglobuline anche da altri punti di vista; esse presentano di regola una minore avidità per l'antigene corrispondente di quanto si verifichi per le Ig con la stessa specificità. Anche in questo sistema - anzi, soprattutto in questo sistema - quindi, si constata che l'ontogenesi di ogni singolo clone riassume quella dell'organismo intero che a sua volta, secondo la ben nota legge di Haeckel, ricapitola la filogenesi.
I geni delle Ig offrono inoltre l'esempio più convincente a favore dell'ipotesi che la disposizione degli introni all'interno dei geni strutturali sia correlata in modo molto stretto con la suddivisione in zone anatomo-funzionali discrete (i cosiddetti dominî) delle molecole proteiche da essi codificate: gli introni di regola separano i tratti di DNA che codificano per tratti di catena polipeptidica destinati a costituire dominî diversi nella struttura tridimensionale definitiva della molecola proteica. Questa tendenza, che per alcuni sistemi gene-proteina è poco evidente o perfino dubbia, diventa addirittura lampante nel caso delle Ig: per esempio, i trascritti primari per le catene μ delle IgM di secrezione contengono 5 introni tutti situati in posizioni di chiaro significato funzionale, in quanto separano l'uno dall'altro 6 segmenti di RNA (gli esoni) aventi ciascuno una funzione ben definita.
B. Le tre fonti di variabilità postzigotica tra geni H ed L con regioni V diverse. Sono queste le responsabili della produzione di Ig diverse per idiotipia e specificità anticorpale. La prima, molto cospicua, dovuta alla possibilità di scelta, tra le molte centinaia di geni V, di quale farà parte del gene completo, è responsabile della variabilità localizzata nelle prime due regioni ipervariabili; alla seconda contribuiscono sia la scelta fra i possibili geni J (e D per le VH) sia i modi con cui vengono effettuate la singola (tra VL e J) o le due saldature (quella tra un gene VH e un gene D, e quella tra il gene D saldato a VH e un gene J); la terza consiste nella distribuzione ‛a pioggia' di mutazioni somatiche puntiformi nelle sezioni meno variabili di questi geni.
Naturalmente, la variabilità tra i tipi di anticorpi prodotti da cloni diversi è ulteriormente incrementata dal fatto che i siti combinatori - da cui in definitiva dipende la specificità delle Ig come anticorpi - risultano dall'associazione di una catena H e di una L (v. fig. 16).
C. La sintesi di un gene completo della parte variabile e della parte costante, in grado quindi di produrre catene H o L. Il cosiddetto gene V completo, con la sua grandissima variabilità interclonale generatasi con i meccanismi appena illustrati, non è in effetti un gene strutturale, in quanto da solo non è in grado di produrre alcuna catena polipeptidica. Gli manca ancora - per poter funzionare - di fondersi con un gene per una parte costante (se è un gene VL con un gene CL; e se è un VH con un geneCH, che può essere μ o δ, o uno dei quattro γ o uno dei due α o il gene ε).
Da questo tipo di fusione genica non viene generata nuova variabilità idiotipica: una volta fabbricatosi il proprio gene VL e il proprio gene VH, ogni clone rimane definitivamente impegnato a produrre molecole Ig dalla specificità che deriva loro da questi tratti di catene, cioè anticorpi che riconoscono un certo determinante antigenico. Ma questo impegno a usare sempre lo stesso VH non comporta affatto l'impegno ad associarsi sempre allo stesso dei vari geni CH che si trovano disposti a valle sullo stesso cluster. Al contrario, la regola è che il gene VH si trova in un primo momento, cioè in ogni clone giovane, insieme al tratto di DNA che contiene le regioni μ e δ, formando così un'unità di trascrizione costituita da VH, μ e δ, inframezzata da una serie di introni; in seguito si trova associato a uno dei geni successivi, cioè a uno dei quattro γ o dei due α oppure all'ε, costituendo anche questa volta, con il gene H a cui si è legato, una singola unità di trascrizione.
Anche in questa saldatura VC, responsabile della formazione di un gene completo e pronto a funzionare, come in quelle precedenti tanto in senso cronologico che topografico (si verificano entrambe a sinistra della saldatura VC) che sono responsabili della formazione della sequenza completa specificante la sezione VH, sono coinvolti rimaneggiamenti del DNA, ciascuno dei quali implica la delezione dell'intero tratto di DNA interposto tra i due punti dello stesso cluster che si saldano insieme. Dato questo meccanismo, l'ordine di saldatura V-μδ seguito poi da V-γ (o α o ε) non potrebbe essere invertito, cioè una volta formatosi un gene VHγ, per esempio, non è più possibile che dallo stesso cluster se ne formi uno VHμδ.
D. Possibili utilizzazioni del trascritto primario prodotto dall'unità di trascrizione. Da un unico tipo di molecole di trascritto primario si generano - per effetto di tagli e ricuciture non uguali per tutte le molecole - mRNA diversi e quindi Ig diverse. Valga come esempio il caso meglio studiato, quello della regione Vμδ, ma uno schema analogo è valido anche per gli altri geni.
Si tratta di un'unità di trascrizione molto lunga: nel topo una ventina di Kb. Il trascritto primario o pre-mRNA da essa prodotto, che ha naturalmente la stessa lunghezza, può subire destini diversi e dar luogo a molecole di mRNA differenti che daranno origine a differenti catene polipeptidiche. In particolare possono cambiare da una molecola all'altra i siti di ‛taglio e ricucitura' (o splicing), cioè i tratti di sequenza che vengono rimossi dal pre-mRNA nel corso della sua maturazione a mRNA.
Esistono due tipi di alternative possibili. Da una dipende il tipo di catene, μ o δ, che verranno prodotte, e dall'altra se queste catene saranno secrete dalla cellula come Ig, cioè anticorpi circolanti, o se invece resteranno ancorate alla membrana cellulare.
Se dal pre-mRNA viene rimossa la regione δ ne risulta un mRNA che inizia con il segmento variabile VDJ e prosegue con la sezione che codifica per una catena μ; se, viceversa, essendo stata rimossa la regione μ intermedia, è maturata una molecola di mRNA VDJ-δ, questa produrrà una catena δ. E questa è la prima scelta.
La seconda dipende anch'essa dalle modalità dello splicing, cioè dalle regioni che sono rimosse nel corso della maturazione del pre-mRNA. Per esempio, se da una molecola di pre-mRNA VDJ-μ viene rimossa la sequenza δ, ma insieme alla sequenza μ viene conservata anche una breve sequenza codificante situata circa 1 Kb a valle del segmento μ4, l'mRNA VDJ-μ che ne risulta produce una catena μm, cioè una catena μ destinata a rimanere ancorata alla membrana della cellula. Se invece questo tratto va perduto insieme al tratto δ, l'mRNA risultante produce una catena μs, cioè una catena μ destinata a essere secreta. Rispetto alla μs, nella catena μm mancano gli ultimi 20 residui amminoacidici (i 20 codoni corrispondenti sono stati rimossi insieme all'ultimo introne), sostituiti dalla sequenza specificata dal breve tratto codificante in più. Questa extrasequenza, lunga 41 residui amminoacidici, consta di tre regioni, una idrofilica di 12 residui, una intermedia molto idrofobica di 26 residui e una idrofilica di soli 3 residui, di cui l'ultimo è quello C terminale. Il tratto molto idrofobico è responsabile dell'ancoraggio della catena μ (e quindi dell'intera molecola IgM) alla membrana cellulare, donde il nome di catena μm in contrapposizione a μs. Nel complesso, quindi, l'mRNA per una catena VDJ-μm offre un esempio di differenziazione topologica di una molecola proteica affidata alla struttura di una delle sue catene e quindi, in ultima analisi, alla sequenza codificante del gene strutturale di quella catena; il monomero μ2L2 è costituito da due catene L e due catene μ, ciascuna munita, nella parte iniziale della sua regione V, di una sequenza segnale che fa sì che, una volta ultimata, essa si trovi nel lume del reticolo endoplasmatico. Ma, nel caso delle μm la molecola IgM finale non è destinata a essere secreta (e per questo basterebbe trovarsi nell'anticamera dell'esterno che è il lume del reticolo endoplasmatico, come appunto si verifica per le μs); essa infatti; pur guardando all'esterno della membrana, deve restarvi ancorata funzionando da recettore della cellula che la ha prodotta. Anche questo secondo indovamento è realizzato dalla molecola stessa, essendo implicito nella sua struttura: il lungo tratto idrofobico preterminale rimane imprigionato a tutto spessore nel doppio foglietto fosfolipidico della membrana cellulare e con esso rimane legata alla membrana l'intera molecola con un meccanismo del genere di quello descritto per la glicoforina, che molto verosimilmente è adottato per molte, se non per tutte le proteine integrali di membrana.
A questi due tipi di splicings, che ammettono delle scelte differenziative, si sovrappone un'altra serie di splicings destinati a rimuovere gli introni, passaggio indispensabile all'espressione di qualunque gene strutturale. Questi splicings, a differenza dei precedenti, sono rigidamente programmati, sono cioè eventi obbligati disposti lungo una catena di processi comuni all'espressione di tutti i geni e tratti di DNA che hanno già compiuto le loro scelte differenziative.
E. Aspetti molecolari. Di molti tratti dei tre clusters per le Ig (LK, Lλ e H) si hanno già, o si stanno acquisendo rapidamente, conoscenze dettagliate fino al livello della sequenza nucleotidica; si cominciano così a individuare alcune delle regole da cui dipende l'esatto posizionamento delle regioni destinate a essere saldate insieme o rimosse, e questo tanto a livello di DNA che di RNA. Non è lontano nemmeno il momento in cui saranno chiarite le basi molecolari di fenomeni del tipo dell'esclusione allelica e di altre esclusioni, come quelle per cui ogni clone produce con efficienza catene L o di tipo k o di tipo λ. L'individuazione delle sequenze richieste per i vari tipi di splicings costituisce la premessa per poter cercare, e presumibilmente trovare, gli enzimi capaci di operare quei tagli e ricuciture così precisi che sono alla base degli eventi appena illustrati.
c) Il ruolo dell'antigene nella risposta immunitaria montata specificamente verso di esso.
È adesso il momento di domandarsi come e dove si inserisca l'antigene nella complessa serie di eventi che culmina nella produzione dell'anticorpo corrispondente. È ben noto che l'antigene non si limita a combinarsi con un anticorpo comunque presente in grande quantità, ma svolge un ruolo decisivo nel provocarne la produzione. Possiamo dunque chiederci in che consista questo ruolo causale indispensabile. La risposta a tale domanda è che esso funge da induttore. L'antigene è infatti l'induttore dell'anticorpo, proprio come il lattosio è l'induttore della β-galattosidasi nel sistema classico di Jacob e Monod (v. microbiologia). In entrambi i sistemi l'induttore gioca un ruolo informativo nullo (l'informazione, sotto forma di geni strutturali, per sintetizzare la β-galattosidasi o l'anticorpo, è una proprietà intrinseca della cellula che le conferisce la capacità potenziale - ma solo potenziale - di fabbricare queste proteine) e un ruolo assolutamente fondamentale come effettore, in quanto rende operante la capacità della cellula di produrre la proteina indotta.
Le differenze tra questi due sistemi, identici sul piano formale, sono tuttavia numerose. E non potrebbe essere altrimenti: infatti, in un caso si tratta di provocare oppure non provocare la trascrizione di qualche gene strutturale in una cellula batterica, nell'altro di stimolare o meno una tumultuosa moltiplicazione, indi la differenziazione e infine l'esplosiva sintesi di una molecola sotto forma di Ig di secrezione invece che di Ig di membrana da parte di un clone. In quest'ultimo tipo di risposta indotta è coinvolta addirittura la moltiplicazione dell'intera cellula, invece che semplicemente l'espressione di alcuni dei suoi geni. Ma, a guardar bene, ciò non fa che rinforzare l'analogia tra i sistemi inducibili dei Batteri e quelli degli organismi superiori: in un organismo unicellulare, per di più semplice come Escherichia coli, non esiste altro modo di amplificare l'espressione di un gene che quello di fargli produrre molte molecole di mRNA; in un eucariote superiore, invece, il numero di molecole stampo può essere amplificato anche a livello di DNA, per esempio rendendo politenico il tratto di DNA la cui espressione deve essere incrementata preferenzialmente rispetto agli altri geni (v. genetica: Citogenetica). Ma per cellule come l'eritroblasto e il linfocita B (o pre-plasmacellula), che senza bisogno di alcuna amplificazione genica si riempiono completamente di Hb o di Ig, l'unico modo per far aumentare la quantità di queste proteine nell'organismo è il moltiplicarsi, che è qualcosa di più che amplificare il DNA, perché con la moltiplicazione cellulare si amplifica anche tutto quello che serve per far esprimere questo DNA amplificato. L'amplificazione genica senza divisione cellulare è invece necessaria quando non è l'organismo a richiedere una maggiore quantità di prodotto genico, ma la cellula stessa, come nel caso dei geni ribosomiali oppure degli istoni. Del resto, il grande numero di questi geni in tutte le cellule di un organismo non costituisce uno spreco, perché tutte le cellule necessitano di molti ribosomi e di molti istoni; mentre vi sarebbe spreco, e addirittura incompatibile con la vita, se per ogni sostanza destinata a essere prodotta sì in grande quantità ma in un solo tipo di cellula specializzata, fosse necessaria la presenza in tutte le cellule di molte copie dei geni incaricati della sua produzione. È per questo che ogni gamete contiene molti geni per gli istoni e uno solo per la catena β dell'Hb: la regolazione della quantità di Hb che deve essere presente nell'organismo avviene tramite il controllo del numero di globuli rossi, non del contenuto di Hb per globulo rosso. Per quanto importante sia l'Hb, l'organismo non si concede il lusso di avere molti geni β per cellula mentre ne può bastare uno solo. Nel caso delle Ig la situazione, come abbiamo visto, è ancora più estrema: di ogni particolare Ig non c'è nemmeno un gene per gamete e si rimanda la sua sintesi a più tardi. Penseranno i cloni immunocompetenti a procurarsi il gene necessario.
In conclusione, quindi, si possono individuare quattro categorie di geni, tenendo conto dell'espressione differenziale di ciascuno di essi rispetto agli altri geni della stessa cellula (proprietà differenziativa esistente anche in organismi unicellulari come i Batteri) o rispetto allo stesso gene in altre cellule dello stesso organismo pluricellulare complesso: a) geni destinati a esprimersi molto e in tutte le cellule dell'organismo; ve ne sono molti, anche centinaia di copie, per gamete; b) geni programmati a produrre moltissime copie del loro prodotto (che certamente prima o poi risulterà indispensabile all'organismo), ma solo in alcune cellule altamente specializzate. Di ognuno di essi ne esistono una o pochissime copie per gamete. In compenso però l'attività sintetica di queste cellule è monopolizzata al loro servizio in misura tale che un globulo rosso, ad esempio, è poco più che acqua ed emoglobina; c) geni che devono fabbricare relativamente poche copie del loro prodotto specifico, ma in tutte le cellule dell'organismo. Anche questi sono presenti di regola in un'unica copia per gamete. I geni delle classi a) e c) sono i geni adibiti alle ‛faccende domestiche' (gli house-keeping genes degli autori anglosassoni): per quanto elevato possa essere il grado di specializzazione e di differenziazione delle cellule degli organismi superiori, ogni cellula che si rispetti deve accudire a se stessa per le esigenze di ordinaria amministrazione, come la glicolisi, la sintesi delle proprie proteine e acidi nucleici, ecc.; d) geni ciascuno dei quali è molto difficile che dovrà funzionare, sia pure per una sola volta, in tutta la vita dell'organismo, ma nel caso che ciò si verifichi allora dovrà farlo con estrema efficienza, il che è possibile solo in un clone ultraspecializzato di cellule. Si tratta, evidentemente, di geni per le parti variabili delle Ig (il termine ‛ultraspecializzato' si riferisce al fatto che ognuno di questi geni non è semplicemente specializzato a produrre Ig, ma addirittura a produrne un singolo tipo). Ebbene, di ciascuno di questi geni che quasi certamente non dovranno mai funzionare nemmeno in un solo clone, non ne esiste nemmeno una singola copia per gamete; quello che invece esiste già nel genoma dello zigote è tutto l'insieme delle premesse affinché il sistema immunitario possa creare motu proprio uno o più doni con quel gene. Questo sistema che, per quanto ne sappiamo, è l'unico cui è richiesto non solo di eseguire un insieme di istruzioni programmate nel genoma ereditato dallo zigote, ma di inventarne di nuove e poi di eseguirle, è stato infatti dotato di una grande ‛fantasia creativa' (il meccanismo di fusione dei geni V, D e J ecc.) e di meccanismi efficacissimi nel predisporre un altissimo premio, in termini di moltiplicazione cellulare preferenziale, al clone (o ai cloni) che ‛abbia fatto tredici', che sia stato cioè così fortunato da inventare proprio il gene occorrente, quello per l'anticorpo specifico proprio verso l'antigene capitato nell'ambiente interno dell'organismo. È appunto qui che viene fuori il ruolo dell'antigene: il risultato delle partite non influenza la compilazione della schedina vincente - che, oltre tutto, è avvenuta prima - ma è la causa della sua premiazione.
Sui meccanismi con i quali l'antigene riesce a provocare tutta la complessa serie di eventi che culminano nella secrezione di grandi quantità dell'anticorpo corrispondente si hanno conoscenze molto incomplete. Anzi, uno dei problemi centrali dell'immunologia attuale riguarda proprio l'inizio di questa serie di processi, in che modo, cioè, l'antigene avvisi della sua presenza le cellule predisposte a produrre l'anticorpo corrispondente. Si sa che queste cellule, oltre a essere ‛predisposte', hanno ancorato sulla propria membrana cellulare ed esposto all'esterno sotto forma di Ig di membrana l'anticorpo che sono potenzialmente capaci di secernere. Nulla sembrerebbe più logico, quindi, che supporre che questo recettore specifico di membrana si accorga direttamente dell'arrivo dell'antigene corrispondente e che la loro combinazione metta in moto gli eventi successivi della risposta cellulare. E invece, di regola, non è affatto così: l'antigene, pur chimicamente tanto affine al suo anticorpo (anche se di membrana), sembra non sia capace di comunicare con esso altro che tramite una complessa serie di intermediari, che comprende due ulteriori tipi di cellule, i macrofagi e i linfociti T o timociti (v. immunologia e immunopatologia: Immunologia generale; v. sistema reticoloendoteliale). Sembra inoltre che anche questi ultimi, per poter cooperare con il linfocita B, cioè con la cellula che, per mezzo delle sue discendenti, le plasmacellule, produrrà l'anticorpo, debbano riconoscere in modo specifico l'antigene in questione; è stato pertanto postulato - ma non ancora trovato - un recettore specifico nella membrana cellulare dei timociti che sembra essere una delle entità più elusive di tutta la biologia, se si giudica dal numero degli sforzi infruttuosi dedicati alla sua ricerca. A rendere questa rete di interazioni ancora più complicata, ma anche certamente più affascinante, si è aggiunta l'osservazione che i timociti comunicano solo con i linfociti B che hanno il loro stesso fenotipo HLA. E questo fenomeno, insieme alla considerevole omologia tra le sequenze delle catene H delle Ig e quelle degli antigeni HLA, e alla sede dei geni che specificano gli effettori del sistema immunitario (cioè i geni di vari componenti del complemento) tutti addensati nella regione HLA, dà la certezza che deve esistere una stretta relazione filogenetica tra il sistema delle Ig e il sistema HLA (v. trapianti).
È comunque certo che, in un modo o nell'altro, la presenza dell'anticorpo nella membrana della cellula immunitaria a mo' di recettore specifico non può essere casuale: esso deve cioè svolgere un ruolo nel processo di induzione della sua propria sintesi da parte dell'antigene corrispondente.
Una volta che il linfocita B, in un modo o nell'altro, è stato avvisato della presenza dell'antigene corrispondente, ha inizio la fase esecutiva efferente di questa specie di arco riflesso. Essa, naturalmente, comincia con l'amplificazione del DNA (cioè con la moltiplicazione cellulare); a questa segue l'espressione dei geni, cioè la loro trascrizione, intercalata da fenomeni di riordinamento del DNA, per cui la Ig prodotta - sempre la stessa come specificità anticorpale - cambia nel corso della clonogenesi la parte costante della propria catena H, e accompagnata da scelte differenziative con effetti topologici (Ig di membrana o di secrezione) che hanno luogo a livello di pre-mRNA (splicings alternativi). Tutte queste sono conseguenze terminali o preterminali della stimolazione specifica del linfocita B. I meccanismi che li precedono - a cominciare dal più misterioso di tutti, il primo - sono ancora compresi solo in minima parte.

La tab. VI presenta in forma schematica le principali acquisizioni sul sistema genetico preposto alla produzione delle Ig.
d) Considerazioni di carattere generale sul sistema delle Ig.
Il sistema delle Ig appare unico da almeno due punti di vista, quello differenziativo (per i livelli a cui si esplica) e quello adattativo (perché in ogni organismo questo tipo di adattamento supera i limiti scritti nel genoma dello zigote da cui deriva l'organismo stesso).
1. Livello differenziativo. - La differenziazione, per ogni cellula di eucariote superiore, è concepita a livello genico essenzialmente come una scelta di quali geni trascrivere, tra quelli esistenti in quella cellula, e di quando e quanto trascriverli (livello trascrizionale); a questa scelta se ne possono sovrapporre altre a livello della traduzione (se, quando e quanto tradurre ognuno dei particolari tipi di mRNA presenti nel citoplasma). Si dà comunque per scontato che la differenziazione riguardi l'uso e non la struttura del DNA, e che non possano formarsi catene polipeptidiche diverse da trascritti primari uguali, cioè che una volta che un certo gene è stato trascritto, il prodotto proteico dell'RNA non possa essere modulato in senso qualitativo.
Invece, nel sistema genetico delle Ig entrambe queste regole sono completamente disattese: la differenziazione comprende, tra i suoi processi, rimaneggiamenti tanto del DNA che del trascritto primario, che determinano l'attività sintetica potenziale delle cellule di ogni clone immunocompotente e - se arriva l'antigene corrispondente - anche l'attività effettiva.
Il primo evento è quello veramente decisivo: quale anticorpo il clone diventerà capace di produrre. Questa specificità viene fissata una volta per tutte perché è il risultato di un insieme di eventi che, essendo casuali e consistendo in parte in delezioni, sono irreversibili. I geni VH e VL definitivi, risultanti da fusioni di geni scelti a caso e di mutazioni puntiformi, non sono quindi frutto di una scelta preordinata ma piuttosto il risultato di un processo stocastico, la cui probabilità di verificarsi in un particolare clone è piccolissima, mentre è molto elevata quella che si verifichi comunque in un clone qualsiasi, perché il numero dei cloni è molto grande. Alla fine di questa fase i vari doni sono differenziati in un modo unico, nel senso che i geni che possiedono sono diversi gli uni dagli altri e non esistevano nel loro comune progenitore, lo zigote. Questo evento fondamentale, il più importante nella storia di ogni clone immunitario, si verifica a livello di DNA.
Il secondo evento è la scelta di quale gene CH fondere col gene VH fabbricato nella fase precedente per costruire il gene H funzionante e ha un significato diverso da due punti di vista fondamentali. Anzitutto, si tratta di un processo preordinato, invece che casuale, che si ripete con modalità abbastanza costanti in tutti i doni e che - se non fosse per il meccanismo molecolare con cui viene realizzato, consistente in una delezione progressiva di segmenti di DNA - sarebbe perfettamente paragonabile al succedersi dell'espressione dei geni dei clusters globinici. Inoltre in questo caso, a differenza del precedente, l'antigene è certamente molto importante come fattore causale, anche se non sono ancora note le modalità della sua azione. Infine, sebbene i dettagli molecolari della fusione tra il gene VH e i geni CH non siano del tutto chiariti, è comunque certo che essi differiscono considerevolmente da quelli delle fusioni VDJ, che sono programmati in modo da dare origine a ulteriore variabilità a valle dei punti di saldatura.
Una decisione ancora successiva (e anche questa volta si può parlare, come nel secondo evento, di decisione) è cosa fare del trascritto primario. Per esempio, nel caso che il gene completo e funzionante costruito nella fase precedente sia un gene VDJ-μδ, ogni singola molecola di trascritto primario può diventare un μm mRNA o un μs mRNA o un δm mRNA o un δs mRNA (cioè un mRNA che codifichi per una catena μ o δ di membrana o di secrezione). Anche in questo tipo di decisione è certo che l'antigene gioca un ruolo importante.
In tutti i casi questi eventi differenziativi consistono soprattutto in saldature di tratti di sequenze nucleotidiche più o meno lontane con eliminazione dei tratti intermedi (splicings); tuttavia, passando progressivamente da un evento più impegnativo (cioè più gravido di conseguenze per il futuro della cellula, anzi del clone) a uno meno impegnativo (in cui ogni decisione riguarda una particolare molecola di trascritto primario e non pregiudica il destino delle altre), cambia il livello in cui si verifica l'evento: gli splicings miziali sono fra tratti di DNA mentre quelli finali si svolgono nel trascritto primario, cioè in una molecola di pre-mRNA.
2. Livello adattativo. - La peculiarità genetica del sistema immunitario consiste nel fatto che esso è un sistema evolutivo in miniatura. Il repertorio delle proteine che un organismo può produrre è scritto nel suo genoma: quello di un batterio come Escherichia coli nel suo DNA, che comprende qualcosa come tremila geni strutturali, e quello di un mammifero nel genoma dello zigote da cui si è formato, che contiene sì molti più geni strutturali, ma, a quanto sembra, non enormemente di più (il loro numero si pensa sia inferiore a centomila e la grande quantità di DNA in eccesso avrebbe funzioni diverse da quella di specificare sequenze amminoacidiche). Lo spettro di adattabilità in un batterio e di adattabilità più differenziazione di un organismo pluricellulare complesso, se intesi in senso qualitativo, non possono quindi uscire da questi limiti: se una certa cellula batterica è lac-, per esempio per una mutazione nonsenso del gene della β-galattosidasi, essa non può utilizzare il lattosio come fonte di carbonio organico, nemmeno se questo adattamento rappresentasse una questione di vita o di morte, cioè nemmeno se il lattosio costituisse l'unica fonte di nutrimento carbonioso. Ben più ampio è lo spettro, almeno potenziale, di adattamento di una vasta popolazione batterica derivata, per restare nell'esempio di prima, da un singolo batterio lac-: se questa popolazione è veramente molto grande - poniamo: dieci miliardi di cellule batteriche - è quasi sicuro che essa nel suo complesso è capace di adattarsi al lattosio come unica fonte di nutrimento non azotato e che, dopo un certo numero di generazioni cellulari molto stentate, crescerà rigogliosamente in quanto sarà costituita da cellule lac+, come se - intendiamoci, ‛come se' - il lattosio avesse indotto la comparsa del gene lac+ in questi batteri invece di avere semplicemente selezionato e favorito la crescita di quei pochi mutanti lac+ preesistenti nella popolazione al momento in cui il lattosio è diventato l'unica fonte di carbonio organico. Tutto questo è ben noto, ma si vuole qui sottolineare che, anche se le popolazioni di organismi non si adattano all'ambiente altro che con meccanismi darwiniani, ciò non ne limita affatto lo spettro di adattabilità: esso è molto grande, si sarebbe tentati di dire ‛infinito' se questa parola avesse un senso biologico. L'efficienza di adattamento di questa ipotetica popolazione batterica lac- al lattosio dipende naturalmente dal numero iniziale di mutanti lac+, che, a sua volta, dipende dalla frequenza di mutazione, essendo in media tanto più grande quanto più è elevata questa frequenza. Come mai allora le frequenze di mutazione sono così basse? La risposta è molto semplice, oltre che ben nota: siccome la specie batterica non sa in anticipo quali mutazioni le potrebbero tornare utili e quali dannose e l'unica cosa che ha imparato è che quasi tutte le mutazioni sono dannose, o addirittura letali, essa ha raggiunto nel corso dell'evoluzione un ragionevole compromesso tra la necessità di mutare, perché in certi casi è fondamentale disporre di cellule mutanti, e la necessità di mutare poco, perché quasi tutti i mutanti sono svantaggiati. Anzi non è azzardato supporre che i valori delle frequenze di mutazioni siano piuttosto strettamente correlati con il rapporto tra la probabilità che una mutazione casuale sia neutra o addirittura vantaggiosa e la probabilità che sia deleteria o addirittura letale. Anche se l'esperimento di informare una specie batterica che le sarebbe molto utile aumentare la frequenza di mutazione di un particolare gene e accertare se, in seguito a questa informazione, essa riesce a un certo punto ad aumentare preferenzialmente la frequenza di mutazione di quel gene, non è stato mai effettuato, è ragionevole supporre che, a lungo andare, l'evoluzione troverebbe il modo di realizzare questo risultato.
Quello che è sicuro è che qualcosa di fondamentalmente simile la natura lo ha già realizzato con il sistema immunitario.
La grandissima versatilità su base genetica di questo sistema è paragonabile a quella di una popolazione in evoluzione, perché esso è effettivamente una popolazione in evoluzione, tutta contenuta in un singolo organismo e tutta derivata da una singola cellula, come del resto lo è una popolazione batterica originatasi da un solo batterio. Ma in questo caso vi è qualcosa di più: le cellule immunitarie si comportano come se fossero state informate che è bene far mutare (o meglio: produrre nuova variabilità) certi geni, quelli delle Ig, proprio come nell'immaginario esperimento illustrato prima. A un certo momento dello sviluppo dell'organismo, un certo tipo di cellule giunte a una certa fase della differenziazione si dedica attivamente a creare forme alternative nuove, le più svariate, ma di un solo tipo di geni, i geni V, mentre per tutti gli altri geni tutto continua come prima. E così si crea lo spettro di variabilità genetica a livello somatico che può rispondere adeguatamente allo spettro antigenico. Manca poi di spiegare come mai il clone a cui è toccato il compito di costruire un certo anticorpo verso un antigene che sia capitato nell'organismo, e con cui sia quindi venuto a contatto, si moltiplichi tumultuosamente mentre gli altri cloni restano dormienti. Il sistema immunitario è predisposto in modo non solo da ‛mutare' con frequenza del cento per cento nelle regioni V a un certo momento del suo sviluppo, ma anche da conferire un premio selettivo ‛particolarmente allettante' al clone che ha fatto la scelta giusta. La microevoluzione di questo sistema intraorganismico è quindi estremamente rapida: altissima mutabilità seguita da altissimi fattori di selezione (= grandissime differenze di fitness).
È proprio a quest'ultimo livello che cade completamente - ripetiamo: completamente, e per una questione di principio - il parallelismo tra una immaginaria popolazione batterica predisposta a mutare con elevata frequenza in un certo locus e la popolazione delle cellule immunitarie predisposte a mutare con una certa frequenza nelle regioni V. Infatti nel caso della popolazione batterica il premio selettivo viene riscosso dalle cellule che, rispetto all'ambiente in cui si trovano, hanno un genotipo che conferisce - a esse stesse, e non alla popolazione nel suo complesso - un vantaggio diretto, come il sapere utilizzare il lattosio o resistere a un fago o poter fare a meno dell'istidina e così via. E tutto questo si inquadra perfettamente nella famosa frase di Jacob: ‟Il sogno di una cellula è diventare due cellule".
Anche per le cellule immunitarie vale la stessa molla: diventano più numerose quelle che si moltiplicano di più. Ed evidentemente non potrebbe essere altrimenti. Ciò che cambia profondamente è la causa che le fa moltiplicare di più e cioè il fatto di produrre un anticorpo corrispondente a un certo antigene capitato nell'ambiente interno dell'organismo. Ebbene, la motivazione per cui questa, e non altre cellule che producono altri anticorpi, si moltiplichi in modo preferenziale non è egoistica come per le cellule batteriche, ma completamente altruistica: è l'organismo nel suo insieme che ne trae vantaggio, non la popolazione di cellule immunitarie che potrebbe sopravvivere come tale altrettanto bene se tutte le sue cellule fossero lasciate proliferare indipendentemente dal tipo di antigeni circolanti. Il fatto è che questa popolazione di cellule somatiche fa parte di un organismo, per cui la sua evoluzione è subordinata a quella della specie di cui quell'organismo fa parte ed è guidata da essa: attraverso una serie di meccanismi complessi e poco chiari messi in moto dall'antigene ogni cellula di questo sistema è persuasa a diventare altruista, a sostituire cioè al ‛sogno di diventare due cellule' il sogno di far riprodurre l'organismo di cui essa fa parte, anche a costo del proprio sacrificio. Ma, sotto questo aspetto, il sistema immunitario non è affatto diverso da tutti gli altri sistemi di un organismo superiore: nemmeno questo inveterato anticonformista è arrivato al punto di trasgredire a questa che, delle leggi a cui sono soggette le cellule degli organismi pluricellulari, è certo la più generale (vi si sottraggono solo le cellule tumorali, che sono tumorali appunto per questo), tanto che un organismo complesso può essere definito come un insieme di cellule tutte dirette al fine di favorire la riproduzione dell'organismo stesso. Del resto esistono entità biologiche ancora più complesse degli organismi pluricellulari (che sono società di cellule), come per esempio gli alveari, che sono di fatto società di organismi ciascuno dei quali ha per fine ultimo e assolutamente prioritario il mantenimento e l'espansione dell'alveare (o, più precisamente, si comporta come se ...). In questi casi la rinuncia di ognuna delle cellule dei singoli individui della società al suo sogno primordiale ‛di diventare due cellule' è fatta a favore di un organismo che ha, a sua volta, rinunciato al sogno di riprodursi a favore della società di organismi di cui fa parte.
4. Utilizzazione della genetica del sangue a fini teorici e applicativi.
a) Utilizzazioni teoriche.
Delle utilizzazioni teoriche della genetica del sangue, che vanno dalla biologia molecolare alla genetica formale (mappatura dei geni), alla genetica di popolazioni, all'antropologia, alla variabilità genetica e all'evoluzione, si è parlato in varie occasioni. L'unica osservazione troppo importante per essere tralasciata è che - sia pure senza mettere in dubbio il grandissimo valore delle informazioni raccolte attraverso la genetica del sangue - occorre andare molto cauti nel generalizzarle. E questo vale soprattutto per l'antropologia (v. razza) e per la variabilità genetica, in quanto entrambe sono costruite quasi esclusivamente su marcatori del sangue: non possiamo pertanto non porci il problema - e con una certa preoccupazione - dei limiti entro cui è lecito affidarsi a quest'unica fonte di informazioni. Del resto, la validità di questo problema è dimostrata dalla grande diversità esistente tra il grado di diversificazione interrazziale, quale lo si può giudicare dai marcatori dell'antropologia fisica classica, e quello che si osserva appunto sui marcatori ematologici; anzi, nell'ambito di questi ultimi, non sono del tutto coincidenti nemmeno i marcatori biochimici e quelli sierologici.
b) Utilizzazioni pratiche.
Le utilizzazioni pratiche della genetica del sangue sono tanto numerose e importanti da non poter essere passate in rassegna in modo sistematico. Per comodità di esposizione le possiamo classificare in tre grandi classi: applicazioni cliniche, applicazioni farmacogenetiche e applicazioni medico-legali.
Applicazioni cliniche. Le conoscenze raccolte sulla genetica del sangue sono state utilizzate per la diagnosi sia di malattie genetiche (e quindi per l'impostazione di una corretta terapia e prevenzione delle manifestazioni cliniche più gravi associate a certi genotipi, come nel caso di anemie emolitiche indotte da farmaci ossidanti), sia dello stato di portatori per forme recessive (e quindi per prevenire, in alcuni casi, la nascita di individui affetti da malattie genetiche). L'argomento è di tale vastità che esula dall'ambito del presente articolo.
Applicazioni farmacogenetiche. Anche nel campo della farmacogenetica - la scienza che si occupa della base genetica delle differenze individuali nella sensibilità ai farmaci - la genetica del sangue ha fornito alcuni degli esempi più interessanti e significativi, tanto a livello di globuli rossi (anemie emolitiche da farmaci su basi enzimopeniche o emoglobiniche) che del siero (ipersensibilità alla succinildicolina, un miorilassante usato comunemente in anestesiologia, risultante in un prolungamento talvolta di ore, invece che di minuti, dell'apnea indotta da questo farmaco). L'importanza pratica di questi dati risulta evidente se solo si tiene conto della grande frequenza che alcune di queste caratteristiche genetiche raggiungono in certe popolazioni (per es. l'enzimopenia per la G-6-PD, che predispone a crisi emolitiche, e quella per la pseudocolinesterasi, comune negli Eschimesi, responsabile del prolungamento dell'apnea indotta da succinildicolina).
Applicazioni medico-legali. I marcatori genetici del sangue presentano due caratteristiche che li rendono particolarmente utili nella medicina forense; l'immutabilità nel corso dell'intera vita dell'individuo e l'ereditarietà secondo leggi semplici, note e quasi completamente esenti da eccezioni. La prima di queste proprietà li rende particolarmente indicati ai fini dell'identificazione di campioni di sangue di cui si desideri stabilire la provenienza. Infatti, siamo ormai in grado di distinguere un grandissimo numero di caratteri genetici del sangue, il che rende molto probabile che si possa arrivare a escludere l'appartenenza di un campione di sangue a un determinato individuo: basta a tal fine che emerga con certezza anche una sola differenza tra il campione di sangue in esame e il sangue del soggetto X. Nel caso, invece, in cui tutti i caratteri esaminabili siano identici, tanto più se vi sono tra questi caratteri molto rari, risulta praticamente certa la provenienza del sangue esaminato da quel determinato individuo.
La seconda proprietà di questi marcatori, cioè il loro essere trasmessi geneticamente in modo semplice e noto, li rende molto adatti per le diagnosi di accertamento di paternità e di tipo di gemellarità (mono- o dizigotismo), utili quindi non solo per la medicina forense, ma anche per la genetica. Quando i marcatori genetici noti erano pochi si poteva solo arrivare a escludere l'ipotesi di paternità; oggi che la conoscenza dei polimorfismi proteici e dell'HLA (come si vede, si è rimasti comunque nell'ambito del sangue) ha grandemente accresciuto il numero dei marcatori genetici noti, è diventato possibile non solo escludere, ma anche individuare positivamente la paternità.
Bibliografia.
Abelson, J., Butz, E. (a cura di), Recombinant DNA, in ‟Science", 1980, CCIX, 4463.
Bretscher, M. S., Membrane structure: some general principles, in ‟Science", 1973, CLXXXI, pp. 622-629.
Bunn, F. H., Forget, B. G., Ranney, H. M., Human haemoglobins, Philadelphia 1977.
Cao, A., Furbetta, M., Galanello, R., Pirastu, M., Applicazioni attuali e future dei recenti progressi di biologia molecolare delle talassemie, in ‟Prospettive in pediatria", 1980, XL, pp. 307-319.
Cavalli-Sforza, L. L., Evoluzione: La moderna teoria dell'evoluzione, in Enciclopedia del Novecento, vol. II, Roma 1977, pp. 871-884.
Giblett, E. R., Genetic markers in human blood, Oxford 1969.
Harris, H., The principles of human biochemical genetics, Amsterdam 1970 (tr. it.: I principî della genetica biochimica umana, Bologna 19752, 19803).
Lewontin, R. C., The genetic basis of evolutionary change, New York 1974.
Livingstone, F. B., Malaria and human polymorphisms, in ‟Annual review of genetics", 1971, V, pp. 33-64.
Lodish, H. F., Rothman, J. E., The assembly of cell membranes, in ‟Scientific American", 1979, CCXL, pp. 38-53.
Luzzatto, L., Genetics of red cell and susceptibility to malaria, in ‟Blood", 1979, LIV, pp. 961-976.
Modiano, G., Genetically determined quantitative protein variations in man, excluding immunoglobulins, in ‟Atti dell'Accademia Nazionale dei Lincei. Memorie" (serie VIII), 1976, XIII, pp. 53-437.
Modiano, G., Grado e natura della variabilità genetica; sua rilevanza biologica, in Colloquio di genetica di popolazioni, Accademia Nazionale dei Lincei, Roma 1976, pp. 63-116.
Modiano, G., Razza, in Enciclopedia del Novecento, vol. V, Roma 1980, pp. 1032-1051.
Montalenti, G., Evoluzione: L'evoluzionismo nella cultura del XX secolo, in Enciclopedia del Novecento, vol. II, Roma 1977, pp. 865-871.
Mourant, A. E., Köpec, A. C., Domaniewska-Sobczak, K., The distribution of the human blood groups and other polymorphisms, London 1976.
Race, R. R., Sanger, R., Blood groups in man, Oxford 19756.
Roitt, I. M., Essential immunology, Oxford 19742.
Silvestroni, E., Bianco, I., Le emoglobine umane. Biochimica, genetica, popolazionistica, patologia e clinica, Roma 1963.
Silvestroni, E., Bianco, I., Modiano, G., Le talassemie, in Trattato italiano di medicina interna (a cura di P. Introzzi), Roma 1982.
Singer, S. J., Nicolson, G. L., The fluid mosaic model of the structure of cell membranes, in ‟Science", 1974, CLXXV, pp. 720-731.
Watkins, W. M., Biochemistry and genetics of the AB0, Lewis and P blood group systems, in Advances in human genetics, vol. X, New York 1980, pp. 1-136.
Weatherall, D. J., Clegg, J. B., The thalassemia syndromes, Oxford 19722.
Organi emopoietici
SOMMARIO: 1. Generalità sugli organi emopoietici e sulle cellule emopoietiche: a) il problema dell'origine delle cellule del sangue; b) cellule staminali ed elementi precursori; c) orientamenti attuali. □ 2. L'emopoiesi nel periodo fetale ed embrionale. □ 3. Midollo osseo: a) struttura del midollo osseo; b) dimensioni del midollo; c) valutazione globale quantitativa del midollo osseo. □ 4. L'esplorazione del midollo osseo nel vivente: a) storia della biopsia midollare; b) tecniche della medullobiopsia; c) l'esame morfologico qualitativo del midollo osseo; d) utilità della sternopuntura in clinica. □ 5. Elementi emocitopoietici del midollo osseo: a) serie eritropoietica; b) serie granulocitopoietica; c) serie piastrinopoietica; d) plasmacellule; e) monociti. □ 6. Elementi cellulari dello stroma del midollo osseo: a) cellule reticolari; b) macrofagi; c) cellule adipose; d) mastcellule. □ 7. Organi linfopoietici: a) generalità; b) concezioni attuali sulla linfocitopoiesi e classificazione degli organi linfoidi; c) natura e significato della ‛ricircolazione linfocitaria'; d) costituzione anatomo-istologica degli organi linfoidi; e) serie evolutiva linfocitopoietica. □ Bibliografia.
1. Generalità sugli organi emopoietici e sulle cellule emopoietiche.
a) Il problema dell'origine delle cellule del sangue.
Nel sangue circolante si possono distinguere tre classi di cellule: i globuli rossi o eritrociti, i globuli bianchi o leucociti, le piastrine o trombociti. I leucociti costituiscono un gruppo eterogeneo e si possono ulteriormente distinguere in granulociti (neutrofili, eosinofili e basofili), linfociti e monociti. Tutti questi elementi hanno una durata di vita limitata: affinché il loro numero nel sangue rimanga costante è necessario che vengano continuamente sostituiti da parte di cellule giovani. La produzione di tali cellule è circoscritta a determinati organi, definiti per questa loro funzione ‛emopoietici'. Questi organi sono altamente specializzati funzionalmente, sì che in alcuni di essi vengono prodotti soltanto determinati tipi di cellule ematiche: è così possibile distinguere organi emopoietici ‛mieloidi' e ‛linfoidi'.
In questi organi è facile riconoscere alcune serie evolutive cellulari che per le loro caratteristiche morfologiche si possono identificare con gerarchie di elementi precursori diretti delle cellule circolanti: più difficile è invece individuare gli elementi più immaturi dai quali si possano far derivare i vari tipi di elementi precursori e stabilire i rapporti che esistono tra di essi.
Per più di settant'anni vi sono state accese polemiche tra gli ematologi su questo problema dell'origine delle cellule del sangue, sostenendo alcuni (unitaristi) che tutte derivassero da un unico elemento staminale, altri invece (dualisti, trialisti) che esistessero più tipi di elementi capostipiti.
Delle varie teorie unitarie meritano ricordo quella di A. Maximow, che considerava il linfocito quale unico elemento capostipite delle cellule del sangue, e quella di Pappenheim-Ferrata. A. Pappenheim riteneva di aver individuato nel linfoidocito (cellula così denominata perché, con le tecniche di colorazione allora usate, appariva grossolanamente simile al linfocito) l'elemento staminale comune delle cellule del sangue. A. Ferrata descrisse come cellula progenitrice comune un elemento simile al linfoidocito, che denominò emocitoblasto. Secondo la dottrina ferratiana, seguita pressoché unanimemente in Italia, l'emocitoblasto sarebbe derivato da una cellula mesenchimale totipotente, capace di dar origine a tutti gli elementi del connettivo e del sangue, l'emoistioblasto. Tra le altre più diffuse teorie vanno ricordate almeno quelle dualiste di O. Naegeli e di F. R. Sabin, A. C. Doan e R. S. Cunningham, quella trialista di V. Schilling e quella polifiletica di E. Undritz. Naegeli ammetteva l'esistenza di due distinti elementi staminali per gli elementi della serie linfoide e mieloide. Sabin, Doan e Cunningham ritenevano che gli elementi della serie rossa derivassero intravascularmente dagli endoteli dei sinusoidi degli organi emopoietici e che le cellule delle altre serie derivassero indipendentemente da una cellula reticolare situata extravascolarmente in questi organi. La teoria trialista di Schilling postulava l'esistenza, accanto a due distinti elementi staminali, linfoide e mieloide, di un terzo elemento capostipite, di natura reticoloendoteliale, destinato a dar origine al monocito. Undritz riteneva dal canto suo che ogni tipo di cellula del sangue, anche ogni singolo tipo di granulocito, derivasse da un precursore suo proprio.
Attualmente il problema dell'origine delle cellule del sangue viene affrontato non solamente sulla base di individuazioni morfologiche, ma con la valutazione delle caratteristiche funzionali dei vari tipi di cellule ematiche. Da questo punto di vista le cellule circolanti appaiono caratterizzate dal fatto di essere altamente differenziate (per es. i globuli rossi nel trasporto dell'ossigeno e dell'anidride carbonica, i linfociti nell'esecuzione di risposte immunitarie, le piastrine nelle funzioni emostatiche ecc.) e di non essere in grado (a eccezione, come si vedrà, dei linfociti) di proliferare e di autoriprodursi.
b) Cellule staminali ed elementi precursori.
Negli organi emopoietici si riscontrano invece cellule meno differenziate, in buona parte in grado di proliferare. Più precisamente si possono individuare gerarchie di cellule in via di maturazione, identificabili sia morfologicamente sia funzionalmente come precursori diretti degli elementi circolanti: queste cellule appaiono indirizzate in modo irreversibile verso un certo tipo di differenziazione e sono dotate di una capacità proliferativa limitata. Accanto a questi elementi si trovano cellule meno differenziate, ma già orientate verso un certo tipo di evoluzione, non identificate morfologicamente, dotate di capacità proliferative più estese, per quanto abitualmente non più in grado di autorinnovarsi; per tali elementi si sono proposte varie nomenclature: ‟early differentiated cells" (v. Mc Culloch, 1968), ‟elementi precursori" (v. Ebbe e Stohlman, 1965), ‟cellule progenitrici" (v. Moore e Metcalf, 1970). Infine negli organi emopoietici (e anche nel sangue circolante) esistono cellule indifferenziate, dotate di totipotenza evolutiva (capaci cioè di dar origine a tutte le cellule ematiche), in grado di proliferare estesamente e di autorinnovarsi: tali cellule vengono definite ‛cellule staminali' (stem cells).
Sulla base di queste considerazioni gli ematologi sono attualmente concordi nell'accettare la teoria unitaria dell'emopoiesi e nell'identificare nella cellula staminale così funzionalmente definita l'elemento progenitore comune degli elementi del sangue.
L'esistenza di una gerarchia funzionale nell'ambito delle cellule degli organi emopoietici è stata provata da numerose indagini sperimentali, che hanno permesso di classificare questi elementi secondo le loro proprietà biologiche: sulla base di questi studi si è anche delineata una nuova nomenclatura, probabilmente provvisoria, per le cellule emopoietiche.
Il trapianto di cellule di midollo osseo o di milza in animali irradiati con dosi subletali (v. Ford e altri, 1959) ha permesso di dimostrare che le cellule trapiantate sono in grado di ripopolare tutti gli organi emopoietici dell'ospite: accorgimenti particolari (impiego di cellule riconoscibili per il loro peculiare assetto cromosomico) hanno permesso anche di stabilire che tutte le cellule che ripopolano i tessuti emopoietici derivano da un'unica cellula. Particolarmente utile per lo studio di questo elemento si è rivelato anche il metodo della formazione di colonie cellulari da parte di cellule emopoietiche nella milza di roditori irradiati (v. Till e Mc Culloch, 1961). Poiché le colonie che si sviluppano sono costituite da tutti i tipi di cellule del sangue e da elementi in grado di dar origine a loro volta a nuove colonie, le cellule responsabili della loro formazione (CFU: Colony Forming Units), in quanto dotate di pluripotenza evolutiva e della capacità di autorinnovarsi, vengono correntemente identificate con le stem cells. Si ritiene generalmente (per quanto le indagini più recenti indichino che le CFU non costituiscono una popolazione omogenea) che gli elementi staminali si trovino in una fase di quiescenza, al di fuori del ciclo cellulare.
L'impiego in varie situazioni sperimentali di una sostanza presente nel plasma in grado di stimolare la formazione dei globuli rossi, l'eritropoietina, ha fornito d'altro lato dati probanti l'esistenza di una popolazione di cellule sensibili a questa sostanza (ESC: Erythropoietin Sensitive Cells), differenti con ogni probabilità dalle CFU. Questi elementi non sono più totipotenti ma appaiono già selettivamente avviati verso la differenziazione eritropoietica, sono cioè delle committed stem cells: a differenza delle CFU sono attivamente in ciclo e appaiono dotati di una discreta capacità proliferativa. Le cellule sensibili all'eritropoietina, nell'ambito dell'inquadramento funzionale delle cellule emopoietiche, vengono considerate le ‛cellule progenitrici' degli elementi della serie eritropoietica.
L'esistenza di una seconda categoria di elementi precursori è stata dedotta dallo studio della formazione di colonie in vitro su agar da parte di cellule emopoietiche: in queste condizioni infatti si sviluppano colonie costituite esclusivamente da granulociti, monociti e macrofagi. Pertanto le cellule responsabili della formazione di colonie su agar (CFC: Colony Forming Cells) vengono attualmente identificate con le ‛cellule progenitrici' per gli elementi della serie granulocitopoietica e monocitaria e assimilate funzionalmente alle cellule sensibili all'eritropoietina.
Numerosi dati sperimentali depongono a favore dell'esistenza di ‛elementi progenitori' pure per le cellule della serie piastrinopoietica: queste cellule sarebbero soggette all'influenza di un fattore plasmatico, la trombopoietina, che esplicherebbe un'azione regolatrice analoga a quella dell'eritropoietina.
Per quanto attiene alla serie linfocitopoietica, partendo dal presupposto che i linfociti siano cellule implicate nell'emissione di risposte immunitarie, gli elementi precursori sono stati identificati in cellule non ancora capaci di produrre quantità rilevanti di anticorpi, ma già in grado di dare una risposta anticorpale, la cui esistenza è stata provata da varie metodiche sperimentali in vivo e in vitro.
c) Orientamenti attuali.
Sebbene gli studi più recenti abbiano permesso un inquadramento soddisfacente delle popolazioni cellulari degli organi emopoietici, molti punti richiedono ancora dei chiarimenti.
Uno dei problemi maggiori è costituito dall'identificazione morfologica delle cellule staminali. In passato si riteneva pressoché concordemente che le cellule capostipiti originassero esclusivamente all'interno dei singoli organi emopoietici da elementi di natura mesenchimale (emoistioblasto secondo la dottrina del Ferrata, cellula reticolare secondo gli autori anglosassoni). Quest'ipotesi è stata però smentita da numerose ricerche di cinetica cellulare che negano la possibilità della formazione di cellule staminali a partire dalle cellule reticolari, e dalla constatazione che cellule staminali si possono riscontrare anche nel circolo.
Per lo più si tende oggi a identificare le cellule staminali con i piccoli linfociti e con le cellule linfocitosimili del midollo (che si distinguono dai piccoli linfociti per la struttura leptocromatica del nucleo e per il citoplasma più ampio, basofilo): varie osservazioni morfologiche e ricerche sperimentali tendono però a smentire l'identificazione delle stem cells con i linfociti. La stessa provata eterogeneità delle CFU depone del resto contro l'identificazione delle cellule staminali esclusivamente con i piccoli linfociti.
In secondo luogo, non completamente risolta appare la questione dell'esistenza o meno di una cellula staminale distinta per il sistema linfoide. La maggior parte dei dati sperimentali depone a favore dell'esistenza di un unico elemento staminale, ma sussistono tuttora argomenti contrari a questa ipotesi. Nel campo della patologia umana si è ritenuta a questo riguardo significativa la presenza di un'anomalia cromosomica in corso di leucemia mieloide cronica, consistente nella presenza del cosiddetto cromosoma Ph1 in tutte le cellule del sangue tranne che nei linfociti (v. Whang e altri, 1963), sebbene ciò possa avere un'interpretazione diversa.
Anche la regolazione dei processi differenziativi e proliferativi delle cellule emopoietiche costituisce un argomento tuttora fonte di discussione e di studio; è importante sottolineare che si tende oggi ad attribuire grande importanza alle influenze ambientali cui sono sottoposte negli organi emopoietici le cellule destinate a divenire elementi del sangue. Si ritiene infatti che in questi organi vi siano le basi strutturali per postulare l'esistenza di ‛microambienti' che esercitano in vario modo un'influenza sulle cellule staminali, influenza che può consistere nel determinare le cellule a riprodursi e/o a differenziarsi. Si è anche supposto che compito dei microambienti, altamente specializzati nell'ambito dei singoli organi, sia quello di dare un primo grado di ‛istruzione' a queste cellule: la differenziazione ulteriore dipenderebbe dalla stimolazione da parte di fattori plasmatici (v. fig. 1).
2. L'emopoiesi nel periodo fetale ed embrionale.
È ben noto che l'emopoiesi nell'uomo inizia verso la terza settimana di vita nell'area extraembrionale, sulla parete esterna del sacco vitellino, in corrispondenza di accumuli di cellule mesenchimali denominati isolotti di Wolff e Pander; gli elementi periferici degli isolotti si differenziano in cellule endoteliali mentre quelli centrali danno origine alle ‛cellule primitive del sangue'. Queste si trasformano in ‛eritroblasti primitivi' o ‛megaloblasti'; a questa prima generazione di eritroblasti fa seguito, a partire dalla sesta settimana, quella degli ‛eritroblasti definitivi', simili a quelli dell'adulto. Successivamente l'attività emopoietica si estende al mesenchima diffuso intraembrionale e quindi si circoscrive a livello del fegato e della milza, che cominciano a funzionare a partire dalla sesta e dalla decima settimana di vita. L'attività emopoietica epatica, principalmente normoeritropoietica ma anche granulocito- e megacariocitopoietica, raggiunge il massimo livello al quinto mese, poi decresce gradualmente sino a scomparire all'epoca della nascita e viene sostituita progressivamente dall'emopoiesi midollare e linfoide propria dell'adulto. L'organo linfocitopoietico che per primo entra in attività è il timo; i primi abbozzi dei linfonodi compaiono verso il terzo mese. L'attività splenica diviene esclusivamente linfocitopoietica verso il quinto mese; l'emopoiesi midollare, che inizia già al terzo mese, raggiunge il massimo della propria attività solo poco prima della nascita, all'ottavo mese.
M. A. S. Moore e D. Metcalf (v., 1971) sulla base di numerose ricerche hanno proposto una visione originale dell'emopoiesi embrionale. Questi autori ritengono che la formazione delle cellule staminali avvenga esclusivamente a livello del sacco vitellino, grazie alla trasformazione di una popolazione indifferenziata di cellule mesenchimali: questo fenomeno si verificherebbe una sola volta nell'ontogenesi.
La successiva presenza di un'attività emopoietica in altre sedi sarebbe dipendente dalla colonizzazione da parte di cellule staminali circolanti originate nel sacco vitellino. Queste cellule colonizzerebbero inizialmente il fegato fetale e il tessuto linfoide primitivo: con il declino dell'attività emopoietica nel sacco vitellino, sarebbero le cellule staminali alloggiate nel fegato a provvedere, dopo esser passate in circolo, alla colonizzazione della milza, del midollo osseo e degli organi linfatici primitivi. La cellula staminale viene descritta come una grande cellula indifferenziata, con citoplasma basofilo e nucleoli ben visibili, corrispondente con ogni probabilità alle CFU. La differenziazione di queste cellule sarebbe legata alle influenze esercitate su di esse dagli organi emopoietici, il cui ruolo sarebbe appunto quello di determinare il destino delle cellule staminali immigrate.
Numerose esperienze hanno condotto gli autori citati a sostenere che anche nell'animale adulto sia in atto una migrazione di cellule staminali dal midollo verso i vari organi emopoietici, importante se non essenziale al mantenimento della loro integrità. Solo il midollo osseo sarebbe in grado di automantenere il proprio potenziale di cellule staminali senza dipendere dall'immigrazione dall'esterno. L'emopoiesi viene così concepita come un processo fortemente dinamico, che garantisce da un lato l'esistenza di una grande riserva proliferativa per gli organi emopoietici e dall'altro una grande flessibilità di risposta a richieste funzionali.
Questo concetto, peraltro non da tutti condiviso, rivoluziona le teorie precedenti che consideravano gli organi emopoietici come strutture autonome, nelle quali la produzione delle cellule del sangue dipende da una popolazione fissa di elementi mesenchimali indifferenziati.
3. Midollo osseo.
a) Struttura del midollo osseo.
Nel midollo osseo si distinguono una componente stromale, vasculo-connettivale, e una parenchimale. Lo stroma è costituito da una rete di fibre connettivali e reticolari nelle cui maglie sono accolte le cellule parenchimali emopoietiche. Al reticolo aderiscono cellule in attiva funzione fagocitaria ed elementi indifferenziati con nucleo ovale, chiaro, definiti per la loro posizione cellule reticolari. I vasi, intimamente connessi alle fibre reticolari, svolgono un'importante funzione di sostegno per le strutture midollari negli spazi ossei ove queste sono contenute e la trama di fibre reticolari a sua volta impedisce il collasso della rete sinusoidale. Le arterie nutritizie dell'osso penetrando nella cavità midollare si ramificano in arterie più piccole e quindi in arteriole, dalle quali si dipartono i capillari arteriosi che si trasformano poi in tubuli endoteliali o si continuano in sinusoidi venosi sacciformi: i capillari venosi a loro volta sboccano in formazioni spongiose più o meno ampie anastomizzate tra di loro.
Accurate osservazioni istologiche su materiale prelevato nel vivente mediante il metodo della mielotomia (v. Burkhardt, 1971) permetterebbero di distinguere nel midollo due tipi di circolazione: uno a funzione trofica, affidato al sistema arteriole → anse arteriose dei capillari → anse venose dei capillari → venule; l'altro, intimamente connesso alla funzione emopoietica, facente capo al sistema sinusoidale. Le cellule che costituiscono la parete dei sinusoidi, dette cellule di sponda, sono dotate di capacità fagocitaria e sono connesse direttamente con analoghi elementi dello stroma reticolare: ricerche di microscopia elettronica hanno dimostrato che la parete dei sinusoidi non è sempre continua, il che permette un certo movimento delle cellule in essi contenute verso il parenchima. L. Weiss (v., 1965) ha descritto il midollo come una struttura instabile, costituita da formazioni vascolari che possono modificarsi grazie alla motilità delle cellule che ne costituiscono la parete, in modo che ne risulta garantito il rapido adattamento a svariate richieste funzionali: emopoiesi, regolazione del flusso sanguigno, immissione in circolo di elementi cellulari o, viceversa, sequestrazione di elementi circolanti, ecc. Per esempio, in caso di anemia la parete dei sinusoidi presenta un maggior numero di aperture e permette così una maggiore liberazione di reticolociti; lo stesso fenomeno si verifica per i granulociti dopo stimolazione con opportune sostanze (endotossine).
Le cellule emopoietiche, a qualunque serie appartengano, sono situate sempre extravascolarmente, disposte in cordoni. Studi istologici hanno consentito di dimostrare che la disposizione delle cellule parenchimali attorno ai vasi non è casuale: gli elementi della serie eritroblastica sono situati preferenzialmente attorno ai sinusoidi parenchimali sotto forma di nidi cellulari; le cellule della serie granulocitopoietica più immature sono abitualmente riscontrabili attorno ai vasi, disposte a mo' di manicotto o a ridosso di un seno collettore addossato al trabecolato osseo; le plasmacellule sono in genere in rapporto con i capillari arteriosi.
Il passaggio delle cellule emopoietiche nel sangue avviene mediante diapedesi attraverso la parete dei sinusoidi: ciò è stato dimostrato da osservazioni di microscopia elettronica (Bessis e Breton-Gorius, 1960) non soltanto per le cellule provviste di motilità ameboide (granulociti neutrofili) ma anche per le cellule sprovviste di motilità attiva quali quelle della serie rossa.
Nel midollo osseo non si trovano vasi linfatici: vi sono invece linfociti, sia dispersi sia raccolti in formazioni nodulari costantemente sprovviste di ‛centro germinativo', la cui presenza si apprezza generalmente meglio nei preparati istologici. La quota linfocitaria midollare è particolarmente abbondante nell'infanzia e aumenta nuovamente nell'età senile.
b) Dimensioni del midollo.
Macroscopicamente si possono distinguere nel midollo due settori: il midollo rosso, costituito in massima parte da cellule emopoietiche, e il midollo giallo, costituito da cellule adipose. Alla nascita tutte le ossa contengono midollo rosso, ma col passar degli anni questo si riduce per lasciar posto al midollo giallo. Nell'adulto, solo alcune ossa (costole, vertebre, sterno, epifisi prossimali e strati diafisari superficiali delle ossa lunghe) contengono cellule emopoietiche, mentre in tutte le altre è contenuto midollo giallo.
Per quanto riguarda il peso e il volume del midollo, N. Mechanik (v., 1926) ha determinato valori ponderali compresi tra 1.600 e 3.700 g, con una media di 2.600, di cui circa la metà rosso; G. Wetzel (v., 1927) ha calcolato che nell'uomo maschio giovane il midollo rosso occupa un volume di circa 1.420 ml, e M. Ludwig (citato da Wetzel) ha ottenuto nei calcoli sui volumi valori di 2.915 ml per il midollo totale e di 1.320 ml per il midollo rosso. È importante rilevare che nel vecchio l'estensione totale delle cavità midollari aumenta notevolmente (a 55 anni raggiunge 4.200 ml secondo i calcoli di Ludwig), per cui la riduzione del midollo rosso a favore di quello giallo rilevabile nell'età senile è forse più relativa che assoluta.
Attenendosi ai calcoli di Mechanik, si può dedurre che il midollo osseo in toto rappresenti dal 3,4 al 5,9% del peso corporeo.
c) Valutazione globale quantitativa del midollo osseo.
Nel 1945 Baserga ha sottolineato l'importanza di studiare l'attività emopoietica non solo dal punto di vista qualitativo, ma anche da quello quantitativo. Un'indagine di questo tipo è molto utile agli ematologi, poiché è in grado di fornire un'idea immediata dell'entità delle popolazioni cellulari emopoietiche e dei processi metabolici che si svolgono quotidianamente nel midollo, e di rendere disponibili dati indispensabili ai calcoli di cinetica cellulare.
I primi tentativi di una valutazione quantitativa della massa cellulare midollare risalgono già al 1910: di quell'epoca è infatti lo studio accurato di Lossen che, basandosi sull'esame di sezioni istologiche, ottenne nell'adulto valori compresi tra 600.000 e 1.100.000 cellule per mm3. Successivamente Isaacs (v., 1932) dilacerando frustoli di tessuto midollare ottenne valori oscillanti tra 900.000 e 1.000.000. La Segerdahl (v., 1935), utilizzando materiale prelevato mediante sternopuntura, riscontrò invece valori molto più bassi, compresi tra 10.000 e 250.000 per mm3; occorre tuttavia considerare che con questo metodo le cause d'errore (diluizione con sangue, partecipazione di grasso, sede della puntura ed età del soggetto) sono tali da indurre alcuni autori a considerare di scarso significato i dati così ottenuti. Attualmente si dispone di metodi più moderni, indiretti, basati sul tempo di maturazione degli elementi, sulla durata e sulla frequenza delle mitosi, ecc. e sull'impiego degli isotopi radioattivi. Castoldi (v., 1963) ha sintetizzato i dati disponibili (ottenuti con vari metodi) nella tab. I.
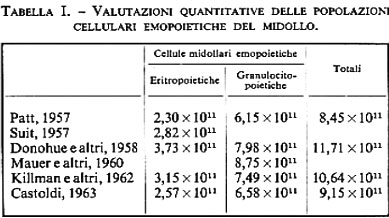
Il numero totale dei megacariociti, secondo Harker (v., 1968), sarebbe di 6,1×106/kg.
Baserga (v., Cyto-démographie..., 1964) ha proposto come dato più probabile per il valore globale delle popolazioni cellulari emopoietiche, per un uomo adulto di 70 kg, quello di 1×1012, e ha calcolato che nel midollo vi siano 7,5×109 cellule in mitosi, il che, per un valore medio di durata delle mitosi di un'ora, equivale a 1,8×1011 mitosi midollari quotidiane.
Questi valori, per quanto necessariamente approssimativi perché molti dei parametri utilizzati per le valutazioni quantitative variano grandemente allo stato normale e molti dati su cui si basa il calcolo non sono del tutto sicuri, permettono tuttavia di affermare che l'entità delle popolazioni midollari è tale da permettere l'applicazione a esse delle leggi della demografia (citodemografia); inoltre il fatto che nel midollo coesistano ingenti popolazioni differenti tra di loro suggerisce l'opportunità di considerarle soggette ai principi dell'ecologia (citoecologia). Il ritmo dei processi mitotici d'altro canto è tale da far ritenere che esistano a livello midollare processi selettivi che implicano l'esistenza di meccanismi di sorveglianza cellulare per il mantenimento dell'omeocitostasi.
4. L'esplorazione del midollo osseo nel vivente.
a) Storia della biopsia midollare.
Nel 1903 Wolff, per la prima volta, impiegò nell'animale da esperimento il metodo della biopsia midollare nel vivente suggerendone la possibile applicazione nella clinica umana; successivamente G. Ghedini praticò la prima medullobiopsia nell'uomo mediante trapanazione ossea in corrispondenza dell'estremità epifisaria prossimale della tibia. Nel 1922 Morris e Falconer impiegarono la biopsia tibiale e la sfruttarono per primi con successo per la coltura del materiale prelevato; nel 1923 Seyfahrt propose la trapanazione ossea dello sterno anziché dell'estremità epifisaria delle ossa lunghe, affinché fosse garantita l'esplorazione di un settore sicuramente attivo del midollo. Sino a questo punto il metodo della medullobiopsia ebbe scarsa applicazione in clinica, soprattutto perché implicava un intervento chirurgico poco gradito al paziente e una minima disponibilità di attrezzature chirurgiche. Solo i pediatri avevano imparato a procurarsi frustoli di midollo con il metodo relativamente incruento della biopsia mediante agopuntura della tibia, possibile solo nei bambini piccoli.
Nel 1929 Arinkin propose di praticare la biopsia sternale nell'adulto mediante un ago, del tipo di quelli che si usano comunemente per la puntura lombare: questo sistema, semplicissimo nella sua esecuzione e ben tollerato dai pazienti, diede rapido impulso allo studio del midollo nel vivente.
b) Tecniche della medullobiopsia.
La biopsia del midollo osseo si può praticare mediante appositi aghi o mediante trivelle che permettono l'estrazione di un cilindretto di tessuto midollare: quest'ultima pratica consente uno studio più accurato, istologico oltre che citologico, del midollo, ed è particolarmente indicata nello studio delle osteopatie e delle mielopatie ipocitemiche (v. Burkhardt, 1971).
L'agobiopsia, che è quella che si pratica più correntemente, si può eseguire in corrispondenza dello sterno, della cresta iliaca e delle apofisi spinose vertebrali. La sede da prediligere nell'adulto è lo sterno: si punge a livello del II-III spazio intercostale, dove lo sterno è ben fissato e abitualmente risparmiato da anomalie congenite. Previa anestesia della cute e del periostio si pratica, mediante un ago, un foro nella cute: si introduce quindi un apposito ago da biopsia, fornito di mandrino, e si perfora il tavolato esterno dello sterno (o il margine di sutura tra teca esterna e interna) infiggendo l'ago con lenti movimenti rotatori in senso perpendicolare (o rispettivamente pressoché parallelo al piano orizzontale). La penetrazione dell'ago nella cavità midollare è svelata dalla sensazione di un brusco venir meno delle resistenze. Si estrae allora il mandrino e si innesta una siringa a perfetta tenuta: si esercita un'aspirazione sufficiente a prelevare circa 0,2 ml di materiale midollare, evitando aspirazioni maggiori che ne provocherebbero un'eccessiva diluizione con il sangue. Il materiale estratto viene raccolto su di un vetrino d'orologio sul quale si è posta una sostanza anticoagulante: i frustoli midollari sono così facilmente individuabili e possono quindi essere raccolti con una pinzetta e giustapposti su vetrini portaoggetti per l'allestimento di preparati citologici; per alcuni è possibile procedere alla fissazione e all'inclusione per le successive ricerche istologiche.
c) L'esame morfologico qualitativo del midollo osseo.
La più semplice delle indagini che si possono eseguire sui preparati midollari è la valutazione della composizione percentuale del midollo (mielogramma): in Italia studi di questo genere iniziarono precocemente, perché si disponeva di una nomenclatura classificativa esauriente quale quella fornita da A. Ferrata. Il mielogramma, per quanto eseguito accuratamente, non rispecchia la reale incidenza percentuale dei vari tipi cellulari del midollo che entro limiti molto ampi. Ciò dipende in parte dalla variabilità della composizione del midollo da un punto all'altro e conformemente all'età del soggetto, in parte dalle modalità di prelievo del materiale. Il metodo della puntura ossea comporta infatti inevitabilmente la commistione con quantità non prevedibili di sangue e la perdita di quelle cellule che, per essere fortemente fissate al tessuto o per le loro dimensioni, sfuggono all'aspirazione: inoltre lo strisciamento del mestruo midollare su vetrino comporta una distribuzione irregolare delle cellule. Va sottolineato anche che per ottenere una valutazione fedele della rappresentazione midollare dei vari tipi cellulari, specie di quelli presenti normalmente in piccola quantità, è necessario esaminare un numero molto grande di cellule. Un'ulteriore causa di discrepanza tra i valori del mielogramma presentati dai vari autori è dovuta al fatto che alcuni ematologi prendono in considerazione nelle loro conte anche i leucociti maturi, che secondo altri vanno invece esclusi per la facilità con la quale il materiale midollare viene commisto a sangue periferico. Per evitare equivoci di questo tipo è stato anche proposto (v. Pontoni, 1936) di denominare la formula midollare che comprende i leucociti maturi non già mielogramma ma ematomielogramma. Sulla base di tali considerazioni, i valori del mielogramma ‛normale' per l'adulto, calcolati da vari autori (v. tab. II), non possono essere considerati che approssimativi.
La composizione citologica del midollo è differente nel bambino rispetto all'adulto: alla nascita si riscontra tipicamente una netta eritroblastosi e il rapporto leuco-eritrogenetico (v. sotto) si aggira attorno all'unità. Già 10-15 gg. dopo la nascita comincia però la prevalenza della serie leucocitaria su quella eritroide e verso i sette anni il rapporto tra le due serie raggiunge i valori propri dell'adulto. Inoltre nel midollo infantile si riscontra un'abbondante quota di linfociti (sino al 10-50%).
Per rendere più facile la lettura del mielogramma e dare una visualizzazione immediata di eventuali deviazioni dalla norma, Baserga nel 1939 ha proposto uno schema per mielogrammi nel quale sono segnate, sotto forma di colonnette tratteggiate, le percentuali che si possono ancora considerare normali per un dato elemento midollare: riportando sullo schema i dati del mielogramma in esame si può immediatamente vedere se esistono variazioni dalla norma per le varie categorie cellulari (v. fig. 2).
Il primo dato che si può ricavare dall'analisi del mielogramma è il cosiddetto indice leuco-eritrogenetico, vale a dire il rapporto tra elementi della serie bianca e della serie rossa. Il rapporto, che va calcolato senza comprendere i granulociti maturi, si aggira normalmente attorno a 2. Di per sé questo indice non ha però grande valore, perché può variare per aumento di uno dei termini o per diminuzione dell'altro e rimane invariato in caso di variazioni simmetriche dei due termini. Molto più importante è invece la valutazione del cosiddetto ‛indice mitotico', cioè della percentuale di cellule in mitosi per tutte le cellule di una data serie maturativa o per ciascuno stadio di una data filiera evolutiva. Già da molti anni numerosi autori si sono occupati della valutazione dell'indice mitotico (IM), che hanno anche impiegato per studi di cinetica cellulare: qui si ricorda soltanto che la quantità globale delle mitosi per tutte le cellule nucleate del midollo ammonterebbe a 6,93× secondo Cronkite e altri (v., 1959) e a 8,86‰ secondo Killman e altri (v., 1962).
d) Utilità della sternopuntura in clinica.
Le modificazioni patologiche della composizione citologica midollare osservabili in corso di processi morbosi possono essere qualitative e/o quantitative. Le modificazioni qualitative più evidenti sono costituite dalla presenza di tipi cellulari abnormi (cellule leucemiche, megaloblasti, cellule di Gaucher, accumuli di cellule neoplastiche, ecc.) o dalla abnorme rappresentazione di alcune cellule nel quadro citologico midollare (per es. plasmacellule in gran numero, disposte a nidi; abbondanza di linfociti). Le anomalie quantitative possono essere relative o assolute: il primo caso si verifica quando vi sia la prevalenza di un determinato tipo cellulare (di elementi immaturi della serie eritropoietica in certe anemie; di eosinofili nei processi allergici e parassitari; di linfociti nella leucosi linfatica cronica, nella malattia di Waldenström, nelle fasi di ripresa dell'agranulocitosi; di elementi reticolari nella cirrosi epatica, nell'endocardite lenta ecc.) o la sua assenza (eritroblastoftisi; agranulocitosi; amegacariocitosi; forme combinate); il secondo caso si realizza in presenza di un'iperplasia di tutte le serie cellulari del midollo (policitemia vera; leucosi mieloide cronica) o un'aplasia globale (atrofia senile; panmielopatie involutive).
Le situazioni patologiche nelle quali è maggiormente utile un esame del midollo sono numerose: il riscontro di una pancitopenia o leucopenia o piastrinopenia periferica, di un'anemia macrocitica o refrattaria o anche ipocromica (se è necessario valutare la disponibilità midollare in ferro); la presenza in circolo di cellule immature o atipiche; il sospetto dell'esistenza di uno stato di discrasia ematica avvalorato dai rilievi semeiologici fisici o radiologici (splenoepatomegalia, linfoadenomegalie, lesioni ossee, ecc.); la sospetta esistenza di condizioni tesaurismosiche o di tumori epiteliali metastatizzati o meno. Quando i rilievi clinici consentono di orientare la diagnosi verso una malattia infettiva o parassitaria (brucellosi, tifo, sepsi, malaria, leishmaniosi, tripanosomiasi ecc.), la biopsia midollare è utile sia per l'identificazione del germe sia per il suo isolamento colturale.
La medullobiopsia è inoltre di grande utilità nella valutazione di alcune condotte terapeutiche, ad es. nei casi in cui sia prevedibile procedere alla splenectomia o quando sia necessario documentare gli effetti determinati sul midollo dalle terapie antianemiche e citostatiche antineoplastiche. Uno degli esempi più felici dell'utilità dello studio del midollo osseo nel vivente è anzi fornito dall'osservazione delle modificazioni indotte dalla terapia antianemico-perniciosa nei soggetti affetti da anemia di Biermer (v. cap. 5, È a).
Un particolare ricordo merita anche la pratica dei tentativi di isolamento di germi patogeni mediante coltura dal midollo osseo che, introdotta sin dall'epoca dei primi tentativi di medullobiopsia (Ghedini, Morris e Falconer, Gerbasi), si è rivelata di utilità diagnostica nel caso di varie malattie infettive. L'impiego di quella che Signorelli ha definito la ‛sternomielocoltura' è particolarmente indicato nella brucellosi e nel periodo ‛muto' del tifo, quando l'emocoltura è già negativa e la sierodiagnosi non è ancora positiva (Baserga e Barbagallo; Debré; Di Benedetto). Il metodo è fruttuoso anche in caso di sepsi (la prima documentazione di fortunata applicazione del metodo si ebbe proprio in corso di sepsi a opera di Morris e Falconer, 1922), come è stato dimostrato anche da Baserga e Barbagallo (v., 1938) mediante l'isolamento di Streptococcus viridans in casi sospetti di endocardite lenta con emocoltura negativa. Talora anche la ricerca di Leishmania nel materiale midollare ha risolto problemi diagnostici.
In questi ultimi anni sta acquistando sempre maggior valore, non solo a scopo di ricerca ma anche di inquadramento clinico, la coltura delle cellule del midollo osseo ottenute per sternopuntura. La coltura del midollo osseo è stata praticata fin dai primi anni di questo secolo, ma solo negli ultimi due decenni se ne è proposto l'uso nello studio dei vari problemi di fisiopatologia midollare, in primis quelli di citocinetica. Generalmente però le cellule midollari sopravviventi in coltura danno precocemente origine a colonie di fibroblasti, vanificando almeno in parte questo tipo di indagini. Ciò non di meno alcune tecniche particolari (colture organotipiche, colture in agar semisolido, colture in coagulo, colture in metilcellulosa) consentono la sopravvivenza, la proliferazione e la maturazione di certi tipi cellulari e hanno permesso di far luce su alcune funzioni del midollo normale e patologico. Così, con la tecnica delle CFU-C (colony forming units in coltura) si possono ottenere tre distinti tipi di colonie, granulocitarie, monocitomacrofagiche e miste, derivate clonalmente da un precursore cellulare non identificato, ma evidentemente preposto alla granulocitopoiesi e alla monocitopoiesi. Analogamente, con la tecnica delle CFU-E (colony forming units sensibili all'eritropoietina) si possono ottenere in vitro colonie eritroidi positive alla reazione alla benzidina. CFU-C e CFU-E deriverebbero da una comune cellula staminale pluripotente, la CFU-S (S sta per spleen, milza), capace di dare origine in vivo a colonie spleniche eritroidi, granulocitarie, megacariocitarie e miste.
Oltre a permettere lo studio delle funzioni dei precursori cellulari midollari, queste tecniche di coltura hanno offerto spunti pratici di discreto significato diagnostico e prognostico. Le CFU-C sono infatti diminuite o assenti nelle leucemie mieloblastiche e linfoblastiche; si normalizzano nelle remissioni e si riducono nuovamente in caso di ricaduta. Nella leucemia mieloide cronica esse risultano molto aumentate, ma si normalizzano per effetto di una terapia adeguata e presentano una marcata diminuzione all'inizio della crisi blastica. Nella mielofibrosi la loro presenza in quantità adeguata risulta di buon significato prognostico, mentre la loro assenza o netta diminuzione costituiscono un signum mali ominis. È stata infine segnalata la loro diminuzione nelle sindromi preleucemiche. Le CFU-C e le CFU-E sono state poi studiate in diverse condizioni sperimentali. È emerso in particolare che esse diminuiscono per effetto di farmaci citostatici e aumentano per effetto degli steroidi, androgeni o meno; quest'ultimo dato potrebbe risultare di grande utilità per la predizione della risposta alla terapia steroidea in varie affezioni mieloaplasiche.
Ricorderemo infine che il puntato sternale offre materiale anche per l'esame cromosomico che può effettuarsi direttamente o dopo arricchimento mediante microcolture colchicinizzate.
5. Elementi emocitopoietici del midollo osseo.
a) Serie eritropoietica.
Nell'ambito di questa sequenza evolutiva, maturazione e divisioni cellulari si succedono con un ritmo definito che comporta un numero determinato di tappe obbligate. Si ammette per lo più che l'evoluzione comporti quattro mitosi e che si possano individuare sei stadi maturativi (proeritroblasto, eritroblasti basofili I e II, eritroblasto policromatofilo, eritroblasto ortocromatico e reticolocito), con la formazione finale di sedici cellule; la capacità di dividersi cessa allo stadio di eritroblasto ortocromatico.
Riguardo alla durata dell'evoluzione eritropoietica, Baserga e Marinone (v., 1944) con lo studio dell'ondata normoblastica che appare nel midollo di soggetti affetti da anemia perniciosa dopo epatoterapia stabilirono che occorrono poche ore per il passaggio da cellula staminale a entroblasto basofilo, 48 ore da eritroblasto basofilo a policromatofilo e 72 da eritroblasto policromatofilo a eritroblasto ortocromatico. Secondo Erslev e Silver (v., 1968) l'evoluzione da proeritroblasto a cellula matura richiede nell'uomo circa 6 giorni; alla fine di questo periodo la cellula, che ha perduto il nucleo, rimane nel midollo ancora 1-2 giorni come reticolocito.
La maturazione degli elementi appartenenti alla serie eritropoietica è caratterizzata dalla progressiva diminuzione delle dimensioni cellulari e dalla modificazione della struttura del nucleo, la cui cromatina tende ad addensarsi in masserelle compatte. La sintesi dell'emoglobina richiede l'integrità del sistema mitocondriale e dell'apparato del Golgi, e la presenza di congrue quantità di ribosomi che conferiscono al citoplasma proprietà basofile (fenomeno paradosso del Ferrata: nel proeritroblasto, destinato a dare origine a cellule contenenti materiale acidofilo quale l'emoglobina, la basofilia citoplasmatica appare aumentata rispetto all'emocitoblasto; v. cellula: Fisiologia della cellula). Successivamente, col progredire della maturazione, la sintesi dell'emoglobina tende progressivamente a diminuire e diminuiscono corrispondentemente i mitocondri e i ribosomi (v. fig. 3).
Il primo elemento individuabile quale appartenente alla filiera evolutiva eritropoietica è il proeritroblasto, descritto da Ferrata. È una cellula rotondeggiante, del diametro di circa 20 μ, provvista di un grande nucleo rotondo, con 1-2 nucleoli, la cui cromatina ha l'aspetto di un fine reticolo e mostra una caratteristica tendenza a disporsi in masserelle; il citoplasma, ridotto a un esile margine, appare blu scuro, con un alone più chiaro perinucleare e zone meno tingibili corrispondenti al centrosoma e ai mitocondri.
Le osservazioni di microscopia elettronica hanno dimostrato la presenza in queste cellule di mitocondri, numerosi ribosomi e molecole disperse di ferritina. Nel midollo i proeritroblasti appaiono frequentemente disposti a corona attorno a una cellula reticolare che emette pseudopodi, i quali s'insinuano tra le cellule eritropoietiche. A questa particolare formazione (‛isolotto eritroblastico' secondo M. Bessis, 1958) si è data molta importanza come espressione morfologica di un rapporto di dipendenza trofica delle cellule della serie rossa dalla cellula reticolare.
La tappa successiva è rappresentata dall'eritroblasto basofilo (o, come generalmente oggi si ammette considerando l'esistenza di un'ulteriore mitosi a questo livello, dagli eritroblasti basofili I e II): queste cellule, del diametro di 10-14 μ, mostrano il citoplasma tipicamente azzurro e il nucleo a disegno cromatinico grossolano (pachicromatico) costituito da masserelle triangolariformi scure disposte a raggio di ruota. Nell'eritroblasto policromatofilo per l'inizio della comparsa dell'emoglobina il citoplasma, nei preparati trattati con coloranti del tipo miscela di Romanowsky, assume un colore particolare che varia dal verde azzurro al rosa sporco: talora nella stessa cellula zone basofile si alternano a zone acidofile. Questo elemento misura 8-12 μ di diametro; il nucleo appare più piccolo di quello degli stadi precedenti, denso, ma lascia ancora apprezzare la disposizione radiale delle zolle cromatiniche. La tappa seguente è rappresentata dall'eritroblasto ortocromatico, che meglio sarebbe definire policromatofilo II, perché il suo citoplasma è ancora leggermente policromatofilo rispetto all'eritrocito maturo; il nucleo appare piccolo, picnotico, intensamente tingibile (v. fig. 4).
Da questa cellula, per perdita del nucleo, prendono origine elementi del diametro di 8-10 μm, non ancora nettamente acidofili, nel cui citoplasma, dopo trattamento con particolari coloranti, compare una struttura caratteristica, arrangiata a reticolo, nota agli ematologi come ‛sostanza granulo-filamentosa'. Questa particolare proprietà ha fatto assegnare a tali elementi il nome di ‛reticolociti': sono cellule che rappresentano l'ultima tappa maturativa antecedente all'eritrocito e soggiornano ulteriormente nel midollo per ancora 1-2 giorni prima di essere immessi in circolo.
Le osservazioni di microscopia elettronica hanno dimostrato che i reticolociti sono ancora provvisti di mitocondri e di ribosomi che sono responsabili della lieve policromatofilia al Giemsa e precipitano per effetto dei coloranti basici dando origine alla sostanza granulo-filamentosa.
Non sempre, anche in condizioni fisiologiche, gli eventi della maturazione e dell'estrusione del nucleo procedono simultanei alla maturazione del citoplasma. Si possono così osservare reticolociti con un nucleo picnotico ed eritroblasti basofili sprovvisti di nucleo; alcuni eritroblasti policromatofili di tipo II possono inoltre evolvere direttamente in eritrociti saltando la tappa reticolocitaria. Non tutte le cellule della serie eritropoietica giungono a maturazione; si è calcolato, anche sulla base di studi condotti sul ricambio dei pigmenti ematici, che un 10-15% degli elementi muore nei vari stadi di sviluppo. Questo fenomeno è noto sotto il nome di ‛eritropoiesi inefficace'.
Nel midollo osseo, anche in condizioni perfettamente fisiologiche, l'applicazione della reazione del blu di Prussia per il ferro permette di svelare la presenza di numerosi eritroblasti provvisti di granulazioni di ferro non emoglobinico: questi elementi sono denominati ‛sideroblasti'.
Studi di microscopia elettronica hanno chiarito che i sideroblasti contengono particolari formazioni chiamate siderosomi, costituite da ammassi di molecole di ferritina contenuti in una sostanza di varia densità, circondata o meno da una membrana. La ferritina è presente normalmente in tutti gli eritroblasti, che andrebbero pertanto sempre considerati sideroblasti; questo termine si riserva però agli elementi che ne contengono in quantità tale da essere riconoscibili a livello di microscopia ottica. In condizioni patologiche compaiono invece sideroblasti che differiscono per varie caratteristiche da quelli normali.
Accanto alla filiera evolutiva descritta, nota come normoeritropoietica, se ne conosce un altro tipo, proprio delle cellule eritropoietiche in situazioni patologiche caratterizzate da scarsa disponibilità di fattori necessari alla sintesi del DNA (vitamina B12, acidi folici), cui si dà il nome di linea megalopoietica. Le ricerche moderne hanno dimostrato che l'anomalia del processo eritropoietico corrisponde in tali casi non già alla comparsa di una linea evolutiva differente da quella normale, bensì a un particolare tipo di sviluppo che gli elementi eritroblasticì presentano quando non dispongono di quantità adeguate dei fattori vitaminici citati. L'elemento capostipite dì questa linea evolutiva è il promegaloblasto, cellula del diametro di circa 30 μm: il suo citoplasma è ampio, a contorni non sempre regolari, di colore blu intenso con un alone perinucleare più chiaro e zone meno tingibili corrispondenti al centrosoma e ai mitocondri; il nucleo è grande, situato centralmente, e presenta una struttura cromatinica tutta particolare, delicata, finemente granulare, nel cui contesto sono ben evidenti da 1 a 4 nucleoli di color blu. Il megaloblasto basofilo (o meglio, in analogia con la classificazione adottata per la serie normoeritropoietica, i megaloblasti basofili I e II) misurano 20-25 μm di diametro; il loro nucleo, più piccolo che nello stadio precedente, mostra una struttura cromatinica più grossolana, costituita da granuli disposti a vezzo di perle, che tendono ad addensarsi in noduli (v. fig. 5). Il megaloblasto policromatofilo I, del diametro di circa 15 μm, è contraddistinto dall'iniziale comparsa dell'emoglobina, che conferisce al citoplasma un colore verdastro: in qualche elemento si può già osservare nell'area perinucleare il color arancio proprio del pigmento ematico. La cromatina nucleare è sistemata in un reticolo costituito da masserelle disposte a vezzo di perle, ma il disegno è chiaro per la presenza di larghi spazi interposti tra le maglie, così che ne risulta un aspetto complessivamente giovanile del nucleo rispetto al citoplasma; questo asincronismo maturativo nucleo-citoplasmatico è una delle caratteristiche dell'evoluzione megalopoietica. Il megaloblasto ortocromatico o policromatofilo II, del diametro di circa 10 μm, ha citoplasma arancione e nucleo piccolo, tendente alla picnosi, ma con cromatina non ancora uniformemente addensata come il normoblasto corrispondente; è frequente notare la scomparsa del nucleo a un livello maturativo meno avanzato rispetto a quello del normoblasto e la sua dissoluzione (cariolisi) e frammentazione (carioressi) intracellulare anziché la sua espulsione.
La natura dell'emopoiesi megaloblastica e la sua patogenesi hanno costituito motivo di discussione per molti anni. Il problema fondamentale era quello di stabilire se essa rappresentasse un tipo di evoluzione eritropoietica diverso e autonomo o l'espressione di una deviazione patologica di un'unica filiera evolutiva eritropoietica. Preziose acquisizioni sono state ottenute con lo studio delle modificazioni che intervengono nel midollo di soggetti affetti da anemia biermeriana trattati con principio antianemico-pernicioso. Con questo metodo Baserga e Marinone (v., 1944) hanno dimostrato che entro poche ore dall'inizio della terapia la struttura cromatinica e il rapporto nucleo-citoplasmatico si modificano in senso normoblastico. Gli elementi già maturi (policromatofili e ortocromatici) evolvono conservando le caratteristiche megaloblastiche, quelli meno maturi modificano invece la loro struttura apparendo come elementi intermedi tra megalo- e normoblasti e danno infine origine a cellule indistinguibili dai normali eritrociti. Questo fenomeno (‛normoblastoidizzazione', secondo Baserga) dimostra che la proliferazione megalopoietica non è l'espressione di una filiera autonoma e differente da quella normoeritropoietica, ma piuttosto di un tipo di sviluppo patologico di un'unica linea evolutiva in condizioni di carenza dei fattori necessari per la sintesi degli acidi nucleici (vitamina B12, acidi folici).
Si ritiene infatti che alla base dell'evoluzione megalopoietica stia un'alterazione della sintesi degli acidi nucleici che determina una modificazione del ciclo cellulare; le cellule carenti dei fattori necessari non sono in grado di sintetizzare DNA in quantità sufficiente a permettere l'espletamento della mitosi. L'esistenza di alterazioni biochimiche della sintesi degli acidi nucleici si riflette nella comparsa di tipiche alterazioni stutturali cromosomiche: i cromosomi appaiono più grandi, sottili e meno strettamente spiralizzati che nel normale; si riscontrano anche rotture cromosomiche, contrazione incompleta, distanziamento dei centromeri, aneuploidia (v. genetica: Citogenetica).
b) Serie granulocitopoietica.
Non si è ancora giunti a un accordo definitivo sulle esatte modalità con cui si effetua la granulocitopoiesi, per quanto si siano proposti a questo riguardo vari modelli. Si è generalmente concordi nell'ammettere che l'evoluzione comporti la comparsa di sei tappe maturative riconoscibili morfologicamente in altrettanti elementi: mieloblasto, promielocito, mielocito, metamielocito, granulocito giovanile e granulocito segmentato, dei quali i primi tre sono in grado di proliferare, gli altri sono soggetti esclusivamente a un processo di maturazione. È così possibile distinguere, nella popolazione granulocitopoietica, un compartimento proliferativo e uno maturativo. La discussione è invece ancora aperta a proposito delle modalità di rinnovamento delle cellule di questa serie. Secondo alcuni autori esso dipende dall'attività mitotica di un'unica cellula staminale il cui gettito viene amplificato da successive divisioni in serie a livello del compartimento proliferativo; altri autori invece ritengono che ogni singolo elemento morfologicamente identificato del compartimento mitotico abbia la possibilità di fungere da elemento staminale. Un'altra teoria sostiene infine che tra gli elementi proliferanti solo il mielocito possa fungere da elemento staminale (v. fig. 6).
Anche i dati riguardanti la durata della granulocitopoiesi sono tuttora discordanti: ciò dipende in parte dalla diversità delle tecniche impiegate, in parte dal fatto che non tutti i soggetti umani presi in esame sono in buone condizioni di salute. Cronkite e Vincent (v., 1969) hanno calcolato che il passaggio dalla cellula staminale al sangue richiede circa 13 giorni; secondo I. Boll (v., 1966) il completamento della maturazione richiede 4 giorni; Fliedner e altri (v., 1964) hanno ottenuto per il tempo che intercorre tra lo stadio di mielocito terminale e la comparsa dei primi granulociti (tempo di ‛emergenza dal midollo') valori oscillanti tra 96 e 144 ore (riducibili a 48 ore in caso di necessità).
La maturazione degli elementi appartenenti a questa serie cellulare è caratterizzata dalla comparsa di granulazioni citoplasmatiche e dalla progressiva segmentazione del nucleo.
L'elemento capostipite è il mieloblasto, cellula del diametro di circa 20 μm, rotondeggiante, con citoplasma basofilo caratterizzato da un'area chiara perinucleare, da una certa disomogeneità complessiva che lascia indovinare l'ultrastruttura e dalla presenza delle tipiche granulazioni colorabili metacromaticamente in rosso violetto col Giemsa (granulazioni azzurrofile). Il nucleo è grande, a struttura reticolare fine, un po' indistinta, e contiene 2-5 nucleoli. È praticamente impossibile distinguere morfologicamente i mieloblasti nei tre tipi proneutrofili, proeosinofili e probasofili, anche tenendo conto dei criteri che a tal fine venivano considerati in passato: granulazioni azzurrofile fini e numerose nella cellula indirizzata in senso neutrofilo, più grosse e meno abbondanti in quella proeosinofila, assenza di granulazioni nell'elemento probasofilo e sua colorabilità metacromatica con il blu di toluidina (secondo Undritz e Marinone).
Le osservazioni di microscopia elettronica hanno dimostrato che nel mieloblasto sono presenti formazioni ergastoplasmatiche granulose, numerosi poliribosomi e granulazioni del diametro medio di 800 nm, generalmente omogenee, che sembrano formarsi all'interno dell'apparato del Golgi.
La tappa successiva è rappresentata dal promielocito, elemento caratterizzato dalla comparsa, accanto alle granulazioni azzurrofile, di quelle specifiche della serie cui appartiene. Misura dai 16 ai 27 μm e presenta un nucleo rotondeggiante od ovalare, con disegno cromatinico più grossolano di quello del mieloblasto, nel quale abitualmente è difficile riconoscere dei nucleoli. Il citoplasma è dapprima tenuamente basofilo, poi, col progredire della maturazione, diviene policromatofilo con un'area perinucleare eosinofila dove compaiono le prime granulazioni specifiche; a queste, in varia misura, sono mescolati i granuli azzurrofili, che si presentano di colore (violaceo o rosso vivo) e forma differenti.
Il promielocito neutrofilo contiene granulazioni specifiche piccole, di color marrone chiaro (v. fig. 7); quelle del promielocito eosinofilo sono invece più grandi, di forma sferoidale, e il loro colore varia col progredire della maturazione dal violaceo all'arancione; infine, le granulazioni del promielocito basofilo sono dapprima basofile, poi, come nell'elemento adulto, caratteristicamente metacromatiche al blu di toluidina.
L'ulteriore maturazione conduce alla formazione del mielocito, cellula di 12-18 μm di diametro, con lo stesso rapporto nucleo-citoplasmatico del promielocito. Il nucleo appare ovale o reniforme e la cromatina tende ad addensarsi in masserelle. Il citoplasma, che ha perduto la basofilia e mostra una tinta che va dal grigio chiaro al rosa scuro, contiene pressoché esclusivamente granulazioni specifiche: piccole, fitte, ovalari, omogenee quelle neutrofile; grandi, rotondeggianti quelle eosinofile; grosse, di forma irregolare, violacee quelle basofile. In esso, osservando con attenzione, si possono scorgere ancora delle granulazioni azzurrofile.
L'esame al microscopio elettronico consente di mettere in evidenza la costante presenza di 1-2 nucleoli e di un corpo del Golgi molto sviluppato nel quale si svolge attivamente la granulopoiesi. Sembra che si formino più generazioni di granuli di varie dimensioni, i cui rapporti reciproci non sono stati ancora chiariti.
Dal mielocito deriva il metamielocito, che differisce dal precedente soltanto per le caratteristiche del nucleo, a bastoncino o reniforme con la concavità corrispondente al centrosoma, e a cromatina densa con ammassi ben visibili. Questa cellula rappresenta una vera ‟forma di passaggio", secondo la definizione di Ferrata, verso i granulociti maturi, che ne differiscono solo per la comparsa di strozzature a livello nucleare.
Il granulocito neutrofilo è una cellula del diametro di 12-14 μm, rotondeggiante, il cui citoplasma contiene granulazioni di color marrone chiaro in quantità variabile da elemento a elemento, in genere fini e discretamente regolari. Il nucleo presenta una struttura cromatinica costituita da masserelle scure separate da spazi chiari ed è caratteristicamente segmentato in più lobi (che nel soggetto normale non sono più di 5), disposti in modo da conferirgli un aspetto a lettera S, Y, Z, o a ferro di cavallo: il numero dei lobi cresce coll'aumentare dell'età della cellula.
Il granulocito eosinofilo (v. fig. 8A) misura circa 14 μm di diametro: il nucleo è per lo più bilobato, a bisaccia, con cromatina di aspetto meno grossolano di quello del neutrofilo. Le caratteristiche granulazioni citoplasmatiche misurano 0,5-1,5 μm e sono di color arancione, di forma rotondeggiante o romboidale. Il granulocito basofilo (v. fig. 8B) ha dimensioni ridotte, comprese tra 10 e 14 μm. Il nucleo, difficilmente definibile perché ricoperto dalle grosse granulazioni che stipano la cellula, è di forma irregolare, talora a trifoglio o a quadrifoglio, e non presenta un disegno cr0matinico vero e proprio ma è piuttosto omogeneo. Le granulazioni citoplasmatiche, di color violetto intenso nei preparati colorati col Giemsa e colorabili metacromaticamente con i coloranti tiazinici, hanno forma irregolare e misurano 0,2-1 μm.
Le osservazioni di microscopia elettronica hanno dimostrato che le granulazioni azzurrofile non scompaiono allo stadio di mielocito ma persistono anche nei granulociti. La produzione dei granuli sembra però cessare negli elementi maturi. Nei granulociti neutrofili si notano un apparato del Golgi e alcuni mitocondri, mentre mancano pressoché totalmente le strutture ergastoplasmatiche. Le granulazioni appaiono di forma eterogenea, a manubrio, a grano di riso. Negli eosinofili i mitocondri appaiono più grandi e più numerosi di quelli dei neutrofili, si riscontrano con maggior frequenza strutture ergastoplasmatiche e anche l'apparato del Golgi è più sviluppato. Le granulazioni sono provviste di membrana e contengono alcune formazioni cristalline in una matrice densa. I granulociti basofili presentano un corpo del Golgi piccolo e strutture ergastoplasmatiche molto modeste. Le granulazioni appaiono costituite da una membrana che racchiude particelle di grandezza variabile da granulo a granulo e talora delle figure mieliniche.
In condizioni patologiche, e particolarmente in caso di alterazione della sintesi degli acidi nucleici come nelle deficienze di vitamina B12 o di acidi folici, compaiono delle forme immature gigantesche, spesso prima ancora che si manifestino alterazioni a carico della serie rossa; l'aumento delle dimensioni è particolarmente evidente negli stadi più maturi (Baserga e Gallo, 1941). I granulociti giganti possono anche mostrare un aumento del numero dei lobi nucleari: prendono allora il nome di macropoliciti.
È utile ricordare che non tutte le scuole ematologiche accettano la nomenclatura ferratiana qui impiegata per descrivere la serie maturativa granulocitopoietica. Così la scuola di Naegeli considera come elemento progenitore dei granulociti la cellula mesenchimale mieloide, nettamente distinta da quella linfoide, che dà origine al mieloblasto (corrispondente all'emocitoblasto di Ferrata) dal quale derivano successivamente il mielocito immaturo, il semimaturo e il maturo (corrispondenti rispettivamente al mieloblasto, promielocito e mielocito di Ferrata) e infine il granulocito maturo. Gli autori americani considerano come cellula capostipite il mieloblasto (che viene a corrispondere all'emocitoblasto di Ferrata) dal quale derivano i mielociti A, B e C (assimilabili al mieloblasto, promielocito e mielocito della scuola italiana), il metamielocito e infine i vari tipi di granulociti. E. Undritz ritiene che vi siano dei progenitori distinti per ogni tipo di granulocito: neutrofiloblasto, eosinofiloblasto, basofiloblasto. Da questi elementi si sviluppano i rispettivi promielociti I e II (che corrispondono rispettivamente al mieloblasto e al promielocito ferratiani), i mielociti e poi i metamielociti e i granulociti maturi.
c) Serie piastrinopoietica.
Numerosissime ricerche (osservazioni sulla cellula vivente, indagini di microscopia elettronica, studi immunologici) hanno confermato che le piastrine derivano da cellule giganti del midollo, i megacariociti, come era già stato intuito da J. H. Wright nel 1906.
Secondo le più recenti acquisizioni di cinetica cellulare (v. Ebbe, 1971) si ritiene che i megacariociti derivino dalla cellula staminale attraverso la tappa intermedia di ‛elemento precursore' attualmente non identificato morfologicamente. Gli elementi riconoscibili quali megacariociti presentano un grado variabile di maturità, che permette la loro distinzione in tre gruppi: megacarioblasti, promegacariociti o megacariociti basofili, megacariociti granulosi. Sembra che nessuna di queste cellule sia abitualmente in grado di proliferare (v. Ebbe e Stohlman, 1965), per cui esse vengono considerate come elementi appartenenti a un pool maturativo distinto da quello proliferativo, costituito da cellule staminali e precursori. Caratteristicamente, l'evoluzione verso la differenziazione megacariocitaria comporta un progressivo raddoppio del contenuto in DNA dei nuclei, il che permette di distinguere nell'uomo 5 classi di elementi sulla base del loro grado di ploidia, da 4 nuclei a 64 nuclei (v. fig. 9). I dati di cui si dispone attualmente non permettono di affermare che il raddoppio del contenuto in DNA del nucleo sia sempre seguito da un processo di divisione nucleare con susseguente aumento del numero di nuclei, come sostenuto da alcuni autori. È stato invece accertato (v. Odell e altri, 1965) che il processo di maturazione citoplasmatica è indipendente dall'aumento del contenuto in DNA del nucleo e può aver luogo in corrispondenza di un grado qualunque di ploidia. Per quanto attiene alla durata dell'evoluzione piastrinopoietica, Baserga (v., 1948) sulla base di osservazioni sperimentali (studio della ripresa midollare nel coniglio dopo intossicazione benzenica), cliniche (osservazione degli effetti dell'epatoterapia in soggetti affetti da anemia perniciosa) e di calcoli matematici concludeva che il tempo minimo che può intercorrere tra la cellula staminale e il megacariocito adulto non è certamente superiore ai sette giorni e che il passaggio da megacariocito giovanile a megacariocito maturo può richiedere meno di quattro giorni. Più recentemente Cronkite e altri (v., 1961) hanno confermato che il passaggio da cellula staminale a megacariocito piastrinopoietico richiede circa sette giorni.
Il primo elemento identificabile morfologicamente quale appartenente alla filiera megacariocitaria è il megacarioblasto (v. fig. 10A), cellula di diametro variabile, generalmente compreso tra i 20 e gli 80 μm; il nucleo, grande, rotondeggiante o reniforme, contiene una quantità relativamente scarsa di cromatina, non priva di un disegno caratteristico; il citoplasma, basofilo, non presenta granulazioni. La tappa successiva è rappresentata dal megacariocito basofilo, il cui citoplasma, poco esteso, è intensamente basofilo tranne che in un'area perinucleare corrispondente all'apparato del Golgi, detta area funzionale di Schwartz, dove, talvolta, cominciano a comparire le prime granulazioni azzurrofile; il nucleo appare per lo più gemmante e presenta una struttura cromatinica più densa rispetto al megacarioblasto. In un ulteriore stadio maturativo, il megacariocito granuloso (v. fig. 10B) appare come una grande cellula con citoplasma ampio, perifericamente contornato da un orletto basofilo, zeppo di granulazioni azzurrofile; il nucleo, grande e spesso plurilobato, mostra la cromatina disposta in blocchi. Nelle cellule più mature le granulazioni azzurrofile appaiono riunite in gruppi, separati da bande di citoplasma non granulato: queste formazioni costituiscono le cosiddette figure di campeggiamento piastrinico e preludono all'imminente liberazione delle piastrine dalla cellula madre.
Le osservazioni di microscopia elettronica hanno dimostrato che le granulazioni azzurrofile vengono secrete dalle cisterne dell'apparato del Golgi, che appare ben sviluppato nei megacariociti. La maturazione comporta, oltre alla comparsa delle granulazioni, quella di un sistema di membrane, dette demarcanti, che frazionano il citoplasma in un insieme di strutture tubuliformi disposte attorno al nucleo, probabilmente in comunicazione con l'ambiente extracellulare.
d) Plasmacellule.
Nel midollo normale si riscontra una piccola quantità di plasmacellule, per lo più disposte attorno ai capillari arteriosi. Circa la loro origine, secondo le varie teorie prospettate nel passato questi elementi sarebbero derivati dal linfocito (A. Maximow) o dalle cellule reticolari (P. G. Unna, K. Rohr, ecc.).
Le tecniche attuali di coltura in vitro dei linfociti e lo studio dei processi di ‛trasformazione linfocitaria' hanno permesso di stabilire che i linfociti, attraverso la tappa intermedia di ‛cellula blastica', possono dar origine a elementi capaci di sintetizzare anticorpi, morfologicamente indistinguibili dalle plasmacellule. Anche sulla base di studi immunologici si ammette oggi che le plasmacellule mature rappresentino lo stadio evolutivo finale di cellule derivate dal midollo e differenziatesi verso linfociti destinati a svolgere funzioni immunitarie di tipo umorale, anticorpale (linfociti B secondo la nomenclatura corrente, appartenenti cioè a un sistema immunitario analogo a quello che negli Uccelli dipende dall'influenza della borsa del Fabrizio). Questi linfociti per effetto della stimolazione antigenica si trasformano in ‛cellule blastiche', che danno origine alle plasmacellule. L'evoluzione da linfocito B a plasmacellula si verifica per lo più negli organi linfatici periferici.
L'evoluzione da linfocito B a cellula blastica e quindi a plasmacellula comporta essenzialmente un aumento della quota ergastoplasmatica necessaria alla produzione degli anticorpi. Questa trasformazione è ben apprezzabile con l'ausilio del microscopio elettronico, ma al microscopio ottico, coll'impiego delle comuni colorazioni, è difficile caratterizzare gli elementi progenitori delle plasmacellule. È innegabile però che si possono riscontrare cellule con caratteri giovanili rispetto alla plasmacellula matura, e alcuni autori (v. Mori e Lennert, 1969) hanno proposto di adottare la terminologia di plasmoblasto e proplasmocito per indicare gli elementi intermedi tra cellula blastica e plasmocito maturo.
Il plasmoblasto viene descritto come una cellula di circa 20 μm di diametro, caratterizzata da intensa basofilia citoplasmatica; il nucleo presenta una disposizione cromatinica che ricorda già quella dell'elemento maturo e contiene un nucleolo, peraltro difficilmente apprezzabile senza l'impiego di colorazioni particolari. Il proplasmocito sarebbe identificabile con un elemento le cui caratteristiche citoplasmatiche e nucleari corrispondono a quelle della cellula matura, dalla quale differisce solo per la disposizione centrale del nucleo.
La plasmacellula matura si presenta come un elemento ovale, del diametro massimo di circa 15 μm; il nucleo, disposto in corrispondenza di uno dei poli della cellula e orientato con il suo maggior asse in senso perpendicolare all'asse maggiore della cellula, presenta un disegno cromatinico caratteristico, a zolle grossolanamente quadrangolari, ipercromiche, disposte a raggio di ruota; il citoplasma è intensamente basofilo e presenta un'area chiara perinucleare a forma di mezzaluna, detta arcoplasma (v. fig. 11). Alcuni elementi mostrano la zona periferica del citoplasma di colore rosso-violetto più intenso e sono designati come piasmacellule fiammeggianti di Undritz; altri contengono una o più formazioni citoplasmatiche rotondeggianti, violette, del diametro di 2-3 μm, i cosiddetti corpi del Russel. Raramente in condizioni normali, più frequentemente in stati patologici, si possono individuare plasmacellule contenenti strutture globuliformi di aspetto vitreo che possono giungere a occupare l'intero citoplasma: sono queste le cellule morulari o cellule di Mott.
Le osservazioni di microscopia elettronica hanno evidenziato che la basofilia della plasmacellula è legata alla presenza di numerose vescicole ergastoplasmatiche appiattite, a superficie rugosa, disposte concentricamente attorno al nucleo: esse mancano solo in corrispondenza dell'arcoplasma, occupato dall'apparato del Golgi. All'interno delle formazioni ergastoplasmatiche è possibile osservare materiale secreto, che si ritiene passi poi nelle cisterne golgiane e da qui all'esterno. Il processo secretivo può però avvenire anche con altre modalità. I corpi del Russel appaiono contenuti all'interno delle vescicole ergastoplasmatiche e costituiti da materiale mucopolisaccaridico e proteico. Non tutti gli autori sono d'accordo sulla natura delle inclusioni sferiche presenti nelle cellule di Mott: mentre alcuni (v. Bessis, 1972) le identificano con i corpi del Russel, altri invece (White, Zucker-Franklin) ritengono che esse debbano essere da questi distinte per alcune caratteristiche citochimiche.
e) Monociti.
Nel midollo osseo in condizioni fisiologiche si riscontrano ben pochi monociti. L'origine di queste cellule è stata lungamente discussa: alcuni autori, come Maximow, ne sostenevano la derivazione linfocitaria, altri, come Schilling, quella reticoloendoteliale, mentre la scuola di Naegeli riteneva che derivassero dal mieloblasto.
Numerosi dati sperimentali acquisiti negli ultimi anni sembrano confermare l'origine midollare dei monociti: ad esempio, l'impiego di particolari tecniche citochimiche (v. Leder, 1967) ha dimostrato che la quota monocitaria midollare è superiore a quella dimostrabile con le comuni colorazioni; così pure, i risultati di ricerche autoradiografiche nel ratto (v. Volkman e Gowans, 1965) consentono di affermare che i monociti derivano dal midollo.
Varie ricerche indicano inoltre che i monociti sono legati da rapporti di parentela con i granulociti. Anzitutto questi due tipi di cellule presentano un'analoga sensibilità al cortisolo e un comportamento cinetico molto simile nel corso dei fenomeni riparativi midollari conseguenti ad agranulocitosi. Inoltre, non sono mancati argomenti favorevoli all'ipotesi di una loro origine comune da un unico elemento progenitore: mediante l'impiego di tecniche di coltura di cellule emopoietiche su agar, infatti, è stato possibile dimostrare l'esistenza di elementi in grado di dar origine esclusivamente a granulociti e monociti. Accurati studi istochimici sul corredo enzimatico dei mielociti e dei promielociti (v. Leder, 1967) hanno permesso di formulare l'ipotesi che queste ultime cellule possano essere considerate come i progenitori comuni dei granulociti e dei monociti: si è pertanto supposto che il monocito rappresenti, accanto al mielocito, una forma alternativa di differenziazione del promielocito. Vengono così in ultima analisi confermate le vedute di Naegeli. La teoria di Schilling, che tanto favore ha goduto in passato per l'analogia funzionale tra monociti ed elementi fagocitanti del sistema reticoloendoteliale, sembra doversi oggi interpretare in senso diametralmente opposto: numerose indagini condotte sui tessuti infiammati e sulle cellule di essudato indicano infatti che il monocito circolante è non già una cellula derivata da elementi istiocitari, bensì quello che può essere considerato come il precursore immediato dei macrofagi presenti nei tessuti infiammati.
Il monocito è un elemento di diametro compreso tra 10 e 25 μm, e di morfologia estremamente variabile. Il nucleo, che si cobra in rosso-violetto chiaro col May Grünwald-Giemsa e ha volume assai incostante, può presentarsi rotondeggiante o reniforme o a lettera E o di forma irregolare; la cromatina forma un reticolo le cui strie sono più visibili là dove maggiore ne è l'estensione, con addensamenti in prossimità dei nodi; non si osservano nucleoli. Il citoplasma, ampio e caratterizzato da una basofilia variabile, assume per lo più una tinta grigio-azzurra; vi si notano fini granulazioni azzurrofile, più numerose e meno regolari di quelle riscontrabili nei linfociti. L'esame dei monociti allo stato vivente consente di dimostrarvi la presenza di veli ondulanti come nelle cellule istiocitarie.
Al microscopio elettronico si nota inoltre l'esistenza di uno o due nucleoli, di scarso ergastoplasma e di molti ribosomi. I granuli azzurrofili appaiono densi, omogenei e circondati da una membrana: si ritiene che rappresentino dei lisosomi primari. La superficie della cellula presenta microvillosità e vescicole di micropinocitosi.
6. Elementi cellulari dello stroma del midollo osseo.
a) Cellule reticolari.
Si riteneva un tempo che queste cellule costituissero un sincizio, ma con osservazioni di microscopia elettronica è stato possibile dimostrare che hanno in realtà confini evidenti. Misurano circa 30 μm e nei preparati citologici, separati dalle fibre cui normalmente aderiscono, appaiono grossolanamente poligonali. Il citoplasma ha una tinta lilla e presenta talora dei granuli azzurrofili; il nucleo è grande, rotondeggiante od ovale, chiaro, con la cromatina disposta in un reticolo delicato a maglie regolari: spiccano 1-3 nucleoli, blu, a contorni netti.
Queste cellule non sono abitualmente fagociti attivi: contengono per lo più solo pigmento da usura. Al microscopio elettronico è dimostrabile in esse la presenza di ribosomi, poliribosomi e rari lisosomi primari.
b) Macrofagi.
Sono cellule del diametro di 30-40 μm, di forma ovale o fusata o irregolarmente poliedrica per la presenza di brevi prolungamenti citoplasmatici; il nucleo, rotondeggiante, chiaro, ricorda quello delle cellule reticolari; il citoplasma è tenuamente basofilo con sfumature azzurrine e può contenere granulazioni azzurrofile. Questi elementi sono caratterizzati dalla presenza di vacuoli e di inclusioni cellulari di ogni tipo (detriti cellulari, residui nucleari, microrganismi, ecc.) che ne testimoniano l'attività fagocitaria. È frequente riscontrarne alcuni in via di degenerazione, con citoplasma vacuolizzato, ad aspetto schiumoso (foam cell) e nucleo picnotico.
Allo stato vivente queste cellule emettono larghe, sottilissime membrane a margine libero ondulato che modificano di continuo la loro forma: corpi solidi e gocce di liquido rimangono impigliati nelle pieghe delle membrane e vengono trascinati all'interno del corpo della cellula. Con il microscopio elettronico si dimostra la presenza in esse di numerosi ribosomi, scarso reticolo endoplasmatico e vari lisosomi.
c) Cellule adipose.
Si ritiene generalmente che siano cellule mesenchimali specializzate, per quanto non tutti gli autori siano concordi sulla loro natura. Il grasso compare nel citoplasma sotto forma di piccole gocce tendenti a confluire per dare poi origine a un'unica goccia che distende il citoplasma e lo riduce a un'esile banda periferica in cui, in corrispondenza di una piccola salienza, si nota un nucleo appiattito.
d) Mastcellule.
Questi elementi, variabili per forma - ora ovale o allungata, ora poliedrica - e per dimensioni - il diametro cellulare medio potendo misurare dai 5 ai 25 μm - sono assai rari nel midollo normale, ma aumentano in varie condizioni patologiche: mielopatie involutive, m. di Waldenström, sindromi ipereparinemiche ecc. Il nucleo presenta grosse zolle cromatiniche o appare omogeneo, e non vi si notano nucleoli. Il citoplasma è stipato da grosse granulazioni, del diametro di 0,3-1,5 μm, colorabili metacromaticamente con i coloranti della serie delle tiazine, violacei al Giemsa, e che, a differenza di quelle dei granulociti basofili, non sono solubili in acqua né in alcool e reagiscono positivamente alla reazione di Hale (v. fig. 12).
Al microscopio elettronico le granulazioni appaiono costituite da materiale amorfo e da strutture lamellari concentriche o da formazioni cristalline circondate da una membrana. Numerose ricerche indicano che le mastcellule intervengono attivamente nel metabolismo dell'eparina e dell'acido ialuronico, rappresentano i principali produttori e vettori dell'istamina e di varie altre sostanze, e svolgono un ruolo importante nella flogosi e in numerosi altri processi patologici (v. infiammazione).
7. Organi linfopoietici.
a) Generalità.
Il concetto di linfopoiesi ha subito negli ultimi anni un mutamento profondo a seguito delle nuove conoscenze emerse sulle proprietà biologiche dei linfociti.
In passato si riteneva che queste cellule vivessero per un periodo di tempo molto limitato e che venissero prodotte all'interno degli organi linfoidi secondo una sequenza ordinata e irreversibile di eventi che, a partire da un elemento immaturo, il linfoblasto, conduceva al piccolo linfocito. Quest'ultimo veniva considerato come il prodotto terminale di una serie evolutiva, incapace di proliferare e differenziarsi ulteriormente.
Numerose ricerche di cinetica cellulare hanno invece messo in luce che esistono due popolazioni linfocitarie, costituite l'una da elementi a vita breve, l'altra da cellule che possono vivere più a lungo, nell'uomo anche per anni. La scoperta del fenomeno della ‛trasformazione' linfocitaria (v. Nowell, 1960) ha consentito di dimostrare che il piccolo linfocito è un elemento non già funzionalmente inerte, bensì in grado di evolvere, per effetto di opportune stimolazioni, verso una cellula con caratteri giovanili (cellula ‛blastica') capace di entrare in mitosi e di differenziarsi in più direzioni. Le ricerche immunologiche hanno inoltre chiarito che i linfociti sono cellule responsabili della risposta immunitaria, sia di tipo umorale (legata alla produzione di anticorpi), sia di tipo cellulare (mediata direttamente da cellule effettrici); in occasione dell'incontro con l'antigene specifico questi elementi vanno incontro al fenomeno della ‛trasformazione' e danno origine a una progenie di cellule che intervengono in vario modo nelle reazioni immunitane (v. immunologia e immunopatologia).
Esperimenti di drenaggio continuo del dotto toracico e studi condotti con l'impiego di traccianti radioattivi e di cellule riconoscibili per la presenza di alterazioni caratteristiche (markers) dei loro cromosomi, hanno potuto mettere in evidenza che la maggior parte dei linfociti non trascorre il suo ciclo vitale nel sangue, ma ‛ricircola' più volte tra il sangue e gli organi linfatici periferici. Le popolazioni cellulari degli organi interessati alla ricircolazione non possono pertanto considerarsi costanti, perché vengono continuamente rinnovate da cellule che vi immigrano dal sangue e che possono, per opportune stimolazioni, ‛trasformarsi' in cellule blastiche: queste a loro volta proliferano e danno origine a una progenie di cellule linfoidi.
Sulla base di queste acquisizioni il vecchio concetto di organo linfoide quale sede di produzione sessile dei linfociti è stato abbandonato a favore di un modello diverso, che considera il sistema linfoide nel suo insieme come un'unità dinamica.
b) Concezioni attuali sulla linfocitopoiesi e classificazione degli organi linfoidi.
Sino a qualche decennio fa si riteneva che la maggior parte dei linfociti venissero prodotti a livello dei linfonodi e del tessuto linfoide intestinale e splenico, in corrispondenza di quelle particolari formazioni, note come centri germinativi, costituite da cellule immature circondate da una corona di piccoli linfociti: questi ultimi erano considerati il prodotto dell'attività proliferativa delle cellule centrali. La valutazione dell'attività linfocitopoietica con l'impiego dei radioisotopi ha però dimostrato che in realtà in queste sedi la produzione di linfociti è molto modesta, mentre un'intensa attività linfocitopoietica si svolge nel timo. È stato inoltre chiarito che i cosiddetti centri germinativi non sono costantemente presenti nei tessuti linfoidi e sono anzi sempre assenti nell'embrione e negli animali allevati sterilmente. L'importanza del timo nell'economia della linfocitopoiesi è stata ribadita da esperimenti di asportazione di quest'organo: la timectomia nell'animale neonato (v. Miller, 1961) determina una rilevabile linfopenia e una deplezione delle popolazioni linfatiche dei tessuti extratimici. Poiché numerosi esperimenti condotti con l'impiego dei radioisotopi e di markers hanno consentito di dimostrare direttamente la migrazione di cellule linfoidi dal timo agli altri organi linfocitopoietici, si ammette attualmente che il timo svolga un ruolo centrale nella linfocitopoiesi e che lo sviluppo degli altri organi linfoidi dipenda dall'esportazione di cellule dal timo.
Se i tessuti extratimici non possono essere considerati come i centri primari della linfocitopoiesi, è tuttavia possibile rilevare in essi un grado notevole di attività proliferativa dopo stimolazione con antigeni: in tali condizioni si assiste a un'ipertrofia linfoide, compaiono centri germinativi e si può apprezzare la neoproduzione di numerosi piccoli linfociti.
Sulla base di queste considerazioni, la linfocitopoiesi non viene più considerata come un processo unitario, che si svolge con uguale intensità in tutti gli organi linfopoietici: accanto a una linfocitopoiesi primaria, a regolazione intrinseca, si distingue una linfocitopoiesi secondaria, regolata dalla presenza di materiale antigenico. Il primo di questi due processi si svolge nel timo e ha lo scopo di formare una popolazione di cellule immunologicamente competenti, ‛vergini', destinate agli organi linfoidi dislocati perifericamente nell'organismo; il secondo si verifica nei tessuti extratimici e ha lo scopo di permettere la formazione di cellule immunologicamente attive in modo specifico nei riguardi di un determinato antigene, le quali differiscono quindi funzionalmente da quelle prodotte nel timo.
Numerose ricerche hanno permesso inoltre di concludere che il midollo osseo svolge un ruolo importante nella linfocitopoiesi. Vi sono numerosi argomenti sperimentali che depongono a favore dell'esistenza nel midollo osseo di un pool di cellule staminali che possono differenziarsi in senso linfoide: quest'ipotesi è stata confermata anche clinicamente per il riscontro di miglioramento di affezioni del sistema immunitario dopo trapianti di midollo osseo. Indagini condotte sfruttando tecniche di parabiosi e l'impiego di markers cromosomici hanno dimostrato a loro volta che le cellule destinate a divenire linfociti sono in grado di emigrare dal midollo.
Attualmente, pertanto, sulla base dei dati sperimentali e clinici disponibili e degli studi embriogenetici, si ritiene che le cellule staminali della linea linfocitopoietica risiedano nel midollo osseo e che da qui migrino verso gli organi linfoidi.
Il timo è, nella maggior parte delle specie, il primo organo linfoide a comparire durante l'ontogenesi; in esso gli elementi precursori dei linfociti si riproducono intensamente e subiscono un processo di ‛istruzione' che permette loro di assumere le caratteristiche funzionali di elementi immunologicamente competenti. Si ritiene che quest'organo rappresenti la prima tappa per le cellule che migrano dal midollo, le quali, raggiunto il grado necessario di maturazione funzionale, migrano poi verso gli organi linfoidi dislocati alla periferia.
Non tutti i linfociti dei tessuti extratimici derivano però dal timo; l'asportazione di quest'organo determina infatti la mancata colonizzazione di alcuni determinati settori del tessuto linfoide dei linfonodi e della milza, ma ne risparmia caratteristicamente altri. Ricerche immunologiche hanno dimostrato inoltre che la timectomia determina un difetto selettivo delle difese immunitarie, e precisamente di quelle di tipo cellulare.
Successivi esperimenti condotti negli Uccelli hanno permesso di stabilire che l'asportazione e l'inattivazione funzionale di un altro organo presente in questi animali, la borsa di Fabrizio, determina la mancata colonizzazione di altri distinti settori del sistema linfatico e un difetto selettivo dell'immunità umorale. A. Szenberg ha pertanto prospettato l'ipotesi dell'esistenza di una suddivisione dicotomica della popolazione linfocitaria dal punto di vista della responsabilità immunitaria. Numerose ricerche e osservazioni cliniche su alcune malattie del sistema immunitario, costituenti veri esperimenti della natura, hanno confermato l'esistenza di una simile duplicità funzionale del sistema linfatico anche nei Mammiferi.
Sulla base di tali considerazioni è stata proposta (v. Miller, 1966) una classificazione funzionale degli organi linfoidi, che vengono distinti in primari o centrali e secondari o periferici. Secondo tale concetto gli organi primari sono le sedi nelle quali elementi progenitori dei linfociti, originati nel midollo osseo, vengono indirizzati a differenziarsi verso elementi immunologicamente competenti, mentre gli organi secondari sono centri costituiti nella maniera più opportuna per permettere alle cellule provenienti dagli organi primari di estrinsecare le loro funzioni immunitarie. La distinzione tra i due tipi di organi si basa su varie differenze, embriogenetiche e funzionali. Gli organi primari sono di origine ecto-endodermica, raggiungono il massimo sviluppo nell'età embrionale per poi scomparire nell'età adulta, sono sede di attivi processi riproduttivi indipendentemente da stimolazioni antigeniche e non sono invece sede di fenomeni immunitari esecutivi (produzione di anticorpi). Gli organi secondari invece sono di origine mesodermica, si sviluppano dopo la nascita, sono sede di attività mitotica non spontanea ma dipendente dalla presenza di antigeni; caratteristicamente in essi hanno luogo eventi immunitari operativi.
Negli Uccelli è stato possibile riconoscere due tipi di organi linfoidi primari, il timo e la borsa di Fabrizio, ciascuno dei quali condiziona la presenza di una popolazione linfocitaria con differente competenza immunologica. Nei Mammiferi in genere e nell'uomo in particolare non si è riusciti a dimostrare l'esistenza di un corrispondente anatomico della borsa di Fabrizio. A questo proposito sono state prospettate però varie ipotesi: si è pensato che le funzioni borsali potessero essere svolte dall'appendice, dalle tonsille, oppure dalle placche del Peyer o dal tessuto linfoide intestinale nella sua totalità o anche da questo associato a quello dei cosiddetti centri germinativi della cute (v. Fichtelius e altri, 1970). Particolare favore ha goduto la teoria che sosteneva che questa funzione spettasse alle placche del Peyer: poiché numerosi dati indicano che esse differiscono dagli organi primari, si è anche pensato che queste formazioni linfoidi possano svolgere una funzione duplice (primaria e secondaria), almeno nelle prime fasi dello sviluppo, o che agiscano producendo un fattore di maturazione diffusibile. Altri autori ritengono possibile che le funzioni borsali possano essere state incorporate dal timo nelle fasi precoci dello sviluppo; altri ancora ritengono che la funzione borsale possa essere generalizzata e diffusa nell'organismo.
Varie esperienze (v. Miller e Mitchell, 1969) indicano che nei Mammiferi le cellule progenitrici di quelle formanti anticorpi provengono dal midollo. Pertanto le cellule linfoidi deputate alle difese immunitarie umorali vengono definite nei Mammiferi come cellule di origine midollare, rispetto a quelle di origine timica, o anche (v. Roitt e altri, 1969) come cellule B (appartenenti cioè a un sistema immunitario analogo a quello borsale degli Uccelli) rispetto a quelle T o timodipendenti.
Le cellule sottoposte all'influenza timica vengono ‛istruite' in modo da divenire responsabili della risposta immunitaria di tipo cellulare: questi elementi, definiti anche (Miller) ‛cellule reattive all'antigene' (ARC: Antigen Reactive Cells), in occasione del contatto con l'antigene si ‛trasformano' e danno origine a un clone di cellule immunologicamente attivate in modo specifico verso quell'antigene. Alcune di queste possono agire immediatamente nella reazione immunitaria come elementi effettori diretti o collaborare con le cellule formanti anticorpi nell'esecuzione della risposta immunitaria di tipo umorale; altre invece mantengono la ‛memoria' del contatto con l'antigene e ‛ricircolano' tra i vari distretti linfocitari, pronte a trasformarsi e a proliferare in occasione di un nuovo incontro con l'antigene (memory cells).
Le cellule sottoposte a influenze di tipo borsale, timoindipendenti, di origine midollare, definite anche (Miller) ‛precursori delle cellule formanti anticorpi' (AFCP: Antibody Forming Cells Precursors), si comportano in modo analogo in occasione del contatto con l'antigene, ma danno origine a cellule formanti anticorpi: la formazione di ‛cellule memoria' anche per i linfociti di tipo B è probabile ma ancora discussa. È stato dimostrato che per l'emissione della risposta immunitaria anticorpale è necessaria nella maggior parte dei casi la collaborazione con ARC di origine timica, ma l'esatta modalità e il significato di questa collaborazione non sono stati ancora perfettamente chiariti (v. fig. 13).
c) Natura e significato della ‛ricircolazione linfocitaria'.
Nel 1945 Baserga, valutando il divario esistente tra l'esiguità della massa linfocitaria circolante e l'entità della popolazione cellulare degli organi linfoidi, prospettava l'ipotesi che i linfociti non siano cellule proprie del sangue, ma che vivano solo per una piccola parte nel sangue, sia che si trovino occasionalmente nel sangue per attraversarlo, sia che la grande massa dei linfociti esplichi altrove le sue funzioni.
Numerosi e complessi esperimenti, in primo luogo quelli condotti nel ratto con la tecnica dell'incannulamento del dotto toracico (v. Yoffey, 1957; v. Gowans, 1959), hanno dimostrato da un lato che la produzione giornaliera di linfociti non rende ragione che di una piccola parte del gettito di cellule linfoidi da parte del dotto toracico, che rappresenta la principale via di deflusso delle cellule linfocitarie dagli organi linfatici al sangue; dall'altro che se le cellule provenienti dal dotto toracico dovessero trascorrere esclusivamente nel sangue il loro ciclo vitale, questo, dato il numero di linfociti presenti in circolo, dovrebbe essere brevissimo, contrariamente a quanto in realtà accade. Si è pertanto concluso che i linfociti sono in grado di entrare e uscire più volte dal sangue, di ‛ricircolare' cioè tra il sangue e gli organi linfatici. Il fenomeno della ricircolazione interessa gli organi linfatici periferici: il timo e il midollo osseo sembrano esserne esclusi, per quanto non si sia ancora giunti a risultati definitivi a questo riguardo.
I linfociti interessati nel traffico sono per la maggior parte piccoli linfociti a lunga vita di origine timica; l'ablazione del timo, infatti, determina una netta diminuzione della massa linfocitaria ricircolante. Sembra però possibile che alcuni dei linfociti ricircolanti abbiano origine dall'attività proliferativa degli organi linfoidi periferici, dal momento che negli animali allevati sterilmente si nota una diminuzione delle cellule ricircolanti. Non tutti i linfociti presenti in circolo sono d'altro lato in grado di ricircolare: è stato infatti dimostrato che una quota pari al 10-20 per cento dei piccoli linfociti ematici è esclusa da questo tipo di migrazione.
Le cellule ricircolanti eseguono più volte il percorso sangue→organi linfoidi→linfa→dotto toracico→sangue. Studi morfologici hanno permesso anche di stabilire quali sono le vie anatomiche seguite dai linfociti: la via di passaggio dal sangue ai linfonodi è rappresentata in questi organi da particolari formazioni vascolari, le venule postcapillari, situate nelle regioni medio-profonde della corticale. Sembra (v. Marchesi e Gowans, 1963) che i linfociti migrino non già passando tra le cellule endoteliali, ma attraversando il loro citoplasma. Nella milza non esistono venule postcapillari, e si ritiene che i linfociti penetrino nelle strutture linfatiche per la via del seno marginale, che circonda la polpa bianca là dove questa confina con quella rossa (v. Goldschneider e Mc Gregor, 1968). Le principali ‛aree di traffico' dei linfonodi e della milza sono rappresentate rispettivamente dalle regioni corticali medie e profonde e dall'area periarteriolare, entrambe, come si vedrà, zone timodipendenti.
Il fenomeno della ‛ricircolazione' linfocitaria ha una grande importanza per l'efficienza delle difese immunitarie dell'organismo: esso permette il rapido accorrere di cellule immunologicamente competenti in un punto qualunque dell'organismo minacciato dall'ingresso di un antigene a esso estraneo. Le cellule ricircolanti giunte a contatto con l'antigene sono in grado di proliferare e di dar origine a elementi immediatamente efficaci nella risposta all'antigene o a cellule che serbano la ‛memoria' del contatto: è così possibile amplificare la risposta immunitaria locale all'antigene e tramandare la ‛cifra' dell'incontro ad altre cellule, a loro volta in grado di ricircolare. Numerose ricerche sperimentali indicano che il pool linfocitario ricircolante ha un'influenza di gran lunga superiore a quella della quota linfocitaria sessile nell'esecuzione delle risposte immunitarie. Hall e Morris (v., 1965) hanno dimostrato, ad es., che solo il 4% delle cellule linfoidi provenienti dalla linfa efferente di un linfonodo dopo stimolazione antigenica viene prodotto localmente, mentre il resto deriva da cellule provenienti dal sangue. Esperimenti di perfusione di milze isolate con antigeni (v. Ford e Gowans, 1967) hanno dimostrato che per l'efficienza della risposta anticorpale nella milza è più importante il ritmo di traffico di cellule linfatiche attraverso l'organo che non il numero di linfociti presenti all'interno di esso.
La continua ‛ricircolazione' dei linfociti, oltre a permettere a queste cellule di svolgere le funzioni di vere ‛sentinelle immunitarie', garantisce anche ai vari organi interessati alla ricircolazione la continua disponibilità di materiale nucleare linfocitario che può essere utilizzato da altre cellule a fini metabolici (funzione ‛trefocitica' del linfocito).
Il piccolo linfocito ricircolante può pertanto essere considerato come una vera ‟cellula spora", secondo la definizione di Baserga, che racchiude in sé potenzialità molteplici e riserve metaboliche cedibili in ogni momento a cellule disposte ovunque nell'organismo.
d) Costituzione anatomoistologica degli organi linfoidi.
Nell'uomo, come in tutti i Mammiferi, si distinguono organi linfoidi veri e propri (linfonodi e milza), organi che accanto alle cellule linfoidi contengono una componente epiteliale (organi linfoepiteliali: timo, tonsille e appendice) e agglomerati di cellule linfoidi, disseminati nelle mucose dei vari apparati (dove si possono riconoscere veri follicoli linfoidi, aggregati, come nell'intestino, o solitari) e diffusi nei vari organi, specie in prossimità dei vasi. Caratteristica fondamentale di tutti i tessuti linfoidi è quella di essere costituiti da uno stroma formato da un reticolo di fibre argentofile cui aderiscono cellule reticolari ed elementi in attiva funzione fagocitaria; nelle maglie di questa rete sono contenute le cellule linfoidi. Tra queste si possono distinguere elementi giovanili, cosiddetti linfoblasti e linfociti, che per le loro dimensioni possono essere distinti in grandi, piccoli e medi. Oltre ai linfociti, negli organi linfoidi sono presenti numerose plasmacellule.
I linfonodi sono organi rotondeggianti o a forma di fagiolo, di volume variabile da un grano di miglio a un'oliva, disposti lungo i vasi linfatici. Sono avvolti da una capsula fibrosa che invia all'interno alcuni prolungamenti o trabecole; tra le varie trabecole e tra queste e la capsula è tesa una rete di fibre argentofile cui aderiscono cellule reticolari e macrofagi. I linfonodi presentano una faccia concava, detta ilo, alla quale giungono i vasi sanguigni e dalla quale si dipartono i vasi linfatici efferenti. Istologicamente in questi organi sono riconoscibili una zona periferica, o corticale, e una zona centrale, o midollare. Nella corticale si notano alcune formazioni rotondeggianti, costituite da ammassi linfocitari designati come follicoli linfatici: questi si continuano nella midollare con l'aspetto di formazioni più irregolari, allungate, dette cordoni midollari. Alla periferia del linfonodo, tra follicoli e capsula, le maglie dello stroma si allargano: si viene così a formare una specie di fessura, il seno marginale, delimitata da macrofagi appiattiti detti cellule di sponda e attraversata da fibre reticolari: a questo seno fanno capo i vasi linfatici afferenti. Da qui la linfa prosegue all'interno del linfonodo passando attraverso fessure simili interposte tra i follicoli, i seni dei noduli, e tra i cordoni e le trabecole, i seni dei cordoni, per sfociare infine in uno spazio più ampio, denominato seno terminale, dal quale originano i linfatici efferenti. I follicoli linfatici presentano durata e morfologia incostanti: possono comparire e scomparire e mostrano una notevole varietà di espressioni morfologiche, dall'aspetto di un accumulo compatto di linfociti a quello di una formazione concentrica, con una zona centrale chiara formata da grandi cellule giovani, talora in mitosi, e una periferica scura, a corona, costituita da piccoli linfociti stipati. Quest'ultimo aspetto ha fatto attribuire a queste strutture il nome di ‛centri germinativi' e in passato si riteneva che la produzione di linfociti avesse luogo esclusivamente a questo livello.
Come si è già ricordato, le ricerche più recenti hanno dimostrato che i ‛centri germinativi' sono in realtà formazioni che compaiono soltanto dopo stimolazione antigenica, e non possono più essere considerate la sede principale di produzione dei linfociti. Si è potuto anche chiarire che i linfociti situati nella corona dei centri non possono ritenersi derivati dall'attività riproduttiva delle cellule situate al centro: la maggior parte di essi appartiene invece al pool dei piccoli linfociti ricircolanti a lunga vita.
Maggiori chiarimenti sul significato delle strutture dei linfonodi sono derivati dalle ricerche immunologiche, che hanno dimostrato che in questi organi si possono distinguere due zone funzionalmente differenti: una timodipendente, costituita da cellule implicate nella risposta immunitaria mediata da cellule, e una borsadipendente, formata da linfociti responsabili delle risposte immunitarie anticorpali. La prima zona corrisponde istologicamente alla regione corticale media e profonda (e alla corona dei centri germinativi), la seconda alla parte centrale dei follicoli linfatici, ai cordoni midollari e alle regioni corticali più periferiche.
Dopo una stimolazione antigenica atta a evocare una risposta immunitaria di tipo cellulare, si nota un'iperplasia della prima zona: vi compaiono grandi cellule blastiche e si manifestano fenomeni proliferativi. L'arrivo nei linfonodi di antigeni che provocano una risposta di tipo anticorpale determina invece la comparsa di fenomeni simili nella regione follicolare. Se lo stimolo è intenso e persistente, si assiste alla comparsa di veri ‛centri germinativi': questi compaiono più precocemente e sono più sviluppati durante una risposta di tipo secondario, cioè a sensibilizzazione già avvenuta. Parte delle cellule neoformate muoiono in situ e i loro detriti vengono captati da macrofagi, che vengono così ad assumere una morfologia particolare (macrofagi ‛a corpi tingibili'). I centri germinativi sono strutture instabili che tendono a scomparire col cessare della stimolazione antigenica. Va ricordato che alcune indagini immunologiche (v. Nossal e Ada, 1971) hanno valorizzato l'importanza dell'impalcatura del centro germinativo, costituita da cellule reticolari dendritiche, dimostrando che sulla superficie di queste cellule possono restare aderenti gli antigeni. Si è avanzata l'ipotesi che la continua presenza dell'antigene in questa posizione possa regolare in vario modo la funzione dei follicoli linfatici.
Il tessuto linfatico della milza è rappresentato dalla cosiddetta polpa bianca, che sulle sezioni dell'organo appare come un insieme di isolotti ovoidali, del diametro medio di 0,2-0,7 mm, che spiccano per il loro colorito grigiastro sulla polpa rossa. Queste formazioni linfatiche, dette corpuscoli del Malpighi, appaiono per lo più disposte a manicotto attorno alle arteriole centrofollicolari.
Ulteriori informazioni sulla struttura delle formazioni linfatiche spleniche, delle quali si conosce oggi anche la specializzazione funzionale, si sono ottenute con lo studio della milza dei Roditori.
La polpa bianca appare separata dalla polpa rossa da una zona marginale, costituita da tessuto reticolare lasso, che circonda una struttura vascolare designata come seno marginale, in cui sboccano alcuni rami delle arteriole centrofollicolari. Questa forniazione vascolare, che presenta molte similitudini col seno circolare che circonda i linfonodi, può essere considerata come la via d'ingresso ai tessuti linfoidi splenici degli antigeni presenti nel sangue.
Nell'ambito della polpa bianca si possono ulteriormente distinguere il tessuto linfoide diffuso e i follicoli linfoidi: il primo è disposto immediatamente attorno all'arteriola centrale, ed è noto anche sotto il nome di tessuto linfoide periarteriolare (PAL); i secondi sono generalmente disposti in prossimità dei due poli di quell'ovoide che è il corpuscolo linfatico splenico e presentano una struttura del tutto simile a quella delle corrispondenti formazioni dei linfonodi, con centri germinativi ben evidenti.
La corona periferica di piccoli linfociti dei centri germinativi dei follicoli splenici è per lo più costituita, come nei linfonodi, da piccoli linfociti ricircolanti, a lunga vita.
Anche nella milza, come nei linfonodi, si possono distinguere due zone funzionalmente differenti: l'una, la zona periarteriolare, rappresenta la sezione timodipendente del tessuto linfoide splenico, mentre l'altra, corrispondente alla porzione centrale dei follicoli linfoidi, costituisce la zona borsadipendente.
Il timo, che alla nascita presenta un volume notevole e pesa circa 10-15 g, aumenta considerevolmente sino al secondo anno di età e poi ancora, anche se non proporzionatamente al ritmo accrescitivo dell'organismo, sino alla pubertà, quando raggiunge il peso di 30-40 g. L'organo in seguito diminuisce progressivamente nelle sue dimensioni e s'impoverisce di parenchima, così che nell'adulto è costituito quasi esclusivamente da stroma e grasso. Il timo è diviso in due lobi, ciascuno dei quali è circondato da una capsula da cui si dipartono verso l'interno alcuni setti che suddividono ulteriormente l'organo in lobuli. In ciascun lobulo si distingue una parte scura periferica, la corticale, e una parte centrale più chiara, la midollare: questa, nella parte più profonda, si continua liberamente con quella dei lobuli circostanti. La corticale e la midollare sono costituite entrambe da una rete di cellule epiteliali a nucleo grande e chiaro, nelle cui maglie sono accolte le cellule linfoidi, piccole, con grande nucleo scuro che occupa quasi tutta la cellula: sono questi i timociti. Nella corticale i timociti sono tanto stipati da rendere pressoché invisibile la trama epiteliale, la quale è invece ben visibile nella midollare dove minore è il numero di cellule linfoidi. In questa regione timica sono caratteristicamente presenti i corpi di Hassal, formazioni sferoidali del diametro di 30-100 μm costituite da cellule epiteliali disposte concentricamente attorno a materiale non vivente, probabilmente nuclei in via di degenerazione, e materiale ialino; le cellule più interne possono contenere nel citoplasma sostanze cheratoialinosimili.
Osservazioni di microscopia elettronica hanno chiarito che i timociti differiscono dai piccoli linfociti soltanto per una minor taglia e per dimensioni minori dei loro organuli. Numerosi reperti ultrastrutturali depongono anche a favore della possibile funzione secretiva delle cellule epiteliali timiche (v. Clark, 1966).
Il timo, specialmente nel periodo dello sviluppo, è sede di processi linfocitopoietici molto intensi; l'attività mitotica dei linfociti timici è molto elevata e il ciclo cellulare di questi elementi è il più breve di tutte le cellule emopoietiche.
Studi istologici e osservazioni di microscopia elettronica hanno dimostrato che le cellule che entrano in mitosi sono disposte in stretto contatto con le cellule epiteliali: si è pertanto avanzata l'ipotesi che queste ultime possano esercitare un'influenza eccitomitotica sulle cellule linfoidi adiacenti.
Dal punto di vista dell'attività riproduttiva vi è però una netta differenza tra la corticale, dove i processi mitotici sono molto intensi, e la midollare, dove invece l'attività riproduttiva è scarsa. Per spiegare questo fenomeno si è pensato che la diversità di comportamento possa essere legata a differenze, almeno funzionali, esistenti tra le cellule epiteliali corticali e midollari.
Alternativamente, si è pensato che le cellule linfoidi midollari possano essere diverse da quelle corticali; in realtà molti dati sperimentali sembrano deporre a favore dell'esistenza di una duplicità della popolazione linfocitaria nel timo.
Le ragioni dell'intensa attività mitotica nel timo non sono state ancora definitivamente chiarite: si è peraltro dimostrato che la maggior parte dei linfociti muoiono in situ e che solo alcuni di essi (in quantità molto minore nell'adulto rispetto al neonato) lasciano il timo. Tra le varie teorie proposte per spiegare il fenomeno le più attendibili sembrano quelle di Burnet (v., 1962), secondo le quali l'intensa attivita mitotica è necessaria per la generazione, attraverso un processo di mutazione casuale, della varietà di linfociti immunocompetenti occorrente per rispondere ai vari tipi di stimoli antigenici, e la morte cellulare intratimica è utile all'eliminazione di cellule capaci di reagire contro il proprio organismo (v. immunologia e immunopatologia: Immunologia generale).
e) Serie evolutiva linfocitopoietica.
Le teorie classiche della linfocitopoiesi ammettevano che la produzione dei linfociti a livello degli organi linfatici avvenisse secondo una sequenza evolutiva che a partire da un elemento con caratteri d'immaturità, il linfoblasto, conduceva, attraverso gli stadi intermedi di grande e medio linfocito, al piccolo linfocito. Ricerche morfologiche e indagini condotte con l'impiego di traccianti radioattivi hanno dimostrato che in realtà una sequenza del genere è realmente operante negli organi linfoidi periferici, ma la candidatura del linfoblasto quale cellula capostipite degli elementi linfocitari è stata messa in discussione dalla scoperta delle capacità evolutive del piccolo linfocito; quest'ultima cellula infatti, per effetto di opportune stimolazioni, può dar origine a una sequela evolutiva esattamente inversa. È opportuno pertanto considerare lo schema classico come reversibile, nel senso che il linfoblasto può essere considerato come l'elemento che dà origine al piccolo linfocito tenendo presente però che il piccolo linfocito può ridifferenziarsi dando a sua volta origine al linfoblasto (v. fig. 14).
La teoria del Ferrata riconosceva quale elemento intermedio tra il linfoblasto e il piccolo linfocito il prolinfocito, attribuendo a questa cellula dimensioni inferiori a quelle del piccolo linfocito circolante e ammettendo la sua abituale assenza dal circolo in condizioni fisiologiche e la sua presenza massiccia in caso di leucosi linfatica cronica. Attualmente (v. Baserga, 1969) si ammette che l'elemento descritto dal Ferrata come prolinfocito non rappresenti l'equivalente morfologico della tappa maturativa intermedia tra linfoblasto e piccolo linfocito, ma piuttosto quello del linfocito patologico della leucosi linfatica cronica (linfocito ‛accumulativo', secondo il concetto di W. Dameshek).
Il linfoblasto appare come un elemento di 15-20 μm, rotondeggiante. Il nucleo è rotondo e occupa buona parte della cellula. La cromatina è disposta in masserelle allungate, a disposizione concentrica; sono visibili uno o al massimo due nucleoli, attorno ai quali la cromatina appare addensata a mo' di cercine. Il citoplasma è scarso, intensamente basofilo.
I grandi e i medi linfociti sono cellule del diametro di 10-12 μm. Il nucleo presenta un disegno cromatinico a blocchi sfumati e non mostra abitualmente nucleoli: questi si possono però mettere in evidenza con l'uso di colorazioni particolari o con l'osservazione a contrasto di fase. Il citoplasma può essere più o meno ampio e presenta un grado variabile di basofilia che rispecchia diversi atteggiamenti metabolici funzionali di queste cellule. Nel citoplasma si possono notare alcune granulazioni azzurrofile, i cosiddetti granuli del Wolf-Michaelis, e formazioni vacuolari ben visibili al microscopio a contrasto di fase, designate come corpi del Gall.
Il piccolo linfocito è una cellula del diametro di circa 7 μm (secondo A. Policard vanno considerati ‛piccoli' i linfociti con diametro inferiore agli 8 μm), che appare costituita quasi esclusivamente dal nucleo. Questo, ovale o tutt'al più reniforme, ha una struttura cromatinica caratteristica, a grosse zolle intensamente tingibili. Il citoplasma appare come un esiguo margine che spesso non abbraccia neppure tutta la circonferenza nucleare; può contenere, al pari delle forme più grandi, granulazioni azzurrofile e corpi del Gall.
Le osservazioni di microscopia elettronica hanno permesso di dimostrare che in tutti i linfociti è presente un nucleolo. Nel citoplasma le granulazioni azzurrofile appaiono come formazioni dense circondate da una membrana; sono presenti alcuni mitocondri, mentre l'apparato del Golgi e l'ergastoplasma sono in genere scarsamente rappresentati. In alcuni elementi però l'ergastoplasma appare ben sviluppato, con un reticolo a superficie rugosa, fornito di ribosomi, e anche l'apparato del Golgi appare più esteso: queste cellule intervengono nella sintesi di anticorpi (v. fig. 15).
Per quanto attiene agli aspetti morfologici della ‛trasformazione linfocitaria', dal momento che questo processo consiste in una graduale evoluzione del piccolo linfocito a cellula blastica, ogni tentativo di classificarne le tappe intermedie è necessariamente arbitrario. Semplificando, si può affermare che gli elementi in corso di trasformazione appaiono come cellule di diametro superiore a quello dei linfociti (intorno ai 12 μm), con un nucleo grande provvisto di nucleoli; la cromatina non è più addensata in zolle ma piuttosto punteggiata. Il citoplasma è ampio, basofilo e presenta un alone chiaro perinucleare. Le cellule ‛trasformate' o ‛blastiche' (v. fig. 16) sono note, per la particolare affinità tintoriale del citoplasma, anche sotto il nome di grandi cellule pironinofile: sono elementi di 15-30 μm di diametro, provvisti di un grande nucleo con più nucleoli, spesso disposto a un polo della cellula. Il citoplasma è basofilo, presenta una zona chiara perinucleare e contiene numerosi vacuoli; altre volte è più pallido e presenta una zona giallastra a uno dei poli.
Osservazioni al microscopio elettronico hanno chiarito che la trasformazione comporta un aumento del numero dei mitocondri, un'iperplasia dell'apparato del Golgi, un incremento del reticolo liscio ergastoplasmatico e della quota ribosomiale; risultano inoltre attivati i processi di micropinocitosi.
Bibliografia.
Baserga, A., La rappresentazione grafica del mielogramma, in ‟Haematologica", 1939, XX, pp. 933-946.
Baserga, A., Sulla durata di vita delle cellule del sangue ed emopoietiche, in ‟Bollettino della Società medico-chirurgica di Pavia", 1944, LVIII, pp. 945-956.
Baserga, A., Valutazioni numeriche globali in ematologia e mielogia. Nota I e II, in ‟Bollettino della Società Italiana di Biologia Sperimentale", 1945, XX, pp. 502-507.
Baserga, A., Sulla durata di evoluzione e di vita dei megacariociti e delle piastrine, in ‟Haematologica", 1948, XXXII, pp. 191-204.
Baserga, A., Attualità in ematologia, 2 voll., Roma 1955-1958.
Baserga, A., Cyto-démographie et cyto-écologie. Bilan quotidien des mitoses hémopoïétiques, in ‟Nouvelle revue française d'hématologie", 1964, IV, pp. 469-478.
Baserga, A., L'omeocitostasi. (I meccanismi di mantenimento della costanza cellulare), in ‟Progresso medico", 1964, XX, pp. 329-334.
Baserga, A., Il timo in ematologia. Introduzione generale: timo e linfociti, in Atti del XX Congresso nazionale della Società Italiana di Ematologia, Roma 11-12 aprile 1965, Pavia 1965, pp. 345-356.
Baserga, A., Il problema del prolinfocito, in ‟Progresso medico", 1969, XXV, pp. 542-544.
Baserga, A., Barbagallo, G., Die Sternalpunktion als bakteriologische Hilfsmethode, in ‟Medizinische Klinik", 1938, XXXVII, pp. 178-182.
Baserga, A., Malacarne, P., Castaldi, G., La biologia dei linfociti, in ‟Haematologica latina", 1969, XII, pp. 217-271.
Baserga, A., Marinone, G., Aspetti morfologici del ritorno alla eritropoiesi normoblastica negli anemici perniciosi trattati, in ‟Bollettino della Società medico-chirurgica di Pavia", 1944, LVIII, pp. 1885-1894.
Bessis, M., Cellules du sang normal et pathologique, Paris 1972.
Boggs, D. R., Cinetica dei leucociti neutrofili in condizioni normali e patologiche, in La dinamica dell'ematopoiesi (a cura di J. A. Lo Bue, A. S. Gordon e altri), Roma 1968, pp. 103-141.
Boll, I., Granulocytopoiese unter physiologischen und pathologischen Bedingungen, Berlin-Heidelberg-New York 1966.
Burkhardt, R., Atlante a colori della istopatologia clinica del midollo osseo e dell'osso, Roma 1971.
Burnet, F.M., Role of the thymus and related organs in immunity, in ‟British medical journal", 1962, II, pp. 807-811.
Castoldi, G. L., Sulla valutazione quantitativa del tessuto midollare emopoietico, in ‟Annali dell'Università di Ferrara", 1963, III, 2, pp. 47-66.
Clark, S. L., Cytological evidence of secretion in the thymus, in Ciba symposium on the thymus: experimental and clinical studies (a cura di G. E. W. Wolstenholme e R. Porter), London 1966, pp. 3-30.
Cronkite, E. P., Bond, V. P., Fliedner, T. M., Paglia, D. A., Adamik, E. R., Studies on the origin, production and distribution of platelets, in Blood platelets (a cura di S. A. Johnson e altri), Boston 1961, pp. 494-610.
Cronkite, E. P., Fliedner, T. M., Bond, V. P., Robertson, J. S., Anatomic and physiologic facts and hypotheses about hemopoietic proliferating systems, in Kinetics of cellular proliferation, New York 1959, pp. 1-14.
Cronkite, E. P., Vincent, P. C., Granulocytopoiesis, in ‟Series haematologica", 1969, II, 4, pp. 3-43.
Ebbe, S., The megakaryocyte: maturation and self-renewal, in The platelets (a cura di K. M. Brinkous, R. W. Shermer e F. K. Mostofi), Baltimore 1971, pp. 1-25.
Ebbe, S., Stohlman, F. Jr., Megakaryocytogenesis in the rat, in ‟Blood", 1965, XXVI, pp. 20-35.
Elves, M. W., The lymphocytes, London 19722.
Erslev, A. G. J., Silver, R. K., Studi in vitro sull'eritropoiesi, in La dinamica dell'ematopoiesi (a cura di J. A. Lo Bue, A. S. Gordon e altri), Roma 1968, pp. 44-59.
Ferrata, A. (a cura di), Le emopatie, Milano 19352.
Fichtelius, K. E., Groth, O., Lidén, S., The skin, a first level lymphoid organ?, in ‟International archives of allergy and applied immunology", 1970, XXXVII, pp. 607-620.
Fliedner, T. M., Cronkite, E. P., Killman, S. A., Bond, V. P., Granulocytopoiesis. I. Emergence and pattern of labeling of neutrophilic granulocytes in humans, in ‟Blood", 1964, XXIV, pp. 683-700.
Ford, C. E., Micklem, H. S., Gray, S. M., Evidence of selective proliferation of reticular cell clones in heavily irradiated mice, in ‟British journal of radiology", 1959, XXXII, p. 280.
Ford, W. L., Gowans, J. L., The role of lymphocyte in antibody formation. II. The influence of lymphocytes migration in the isolated perfused spleen, in ‟Proceedings of the Royal Society" (Series B), 1967, CLXVIII, p. 244.
Goldschneider, I., Mc Gregor, D. D., Migration of lymphocytes and thymocytes in the rat. I. The route of migration from blood to spleen and lymphonodes, in ‟Journal of experimental medicine", 1968, CXXVII, pp. 155-168.
Gowans, J. L., The recirculation of lymphocytes from blood to lymph in the rat, in ‟Journal of physiology", 1959, CCXLVI, pp. 54-69.
Hall, J. G., Morris, B., The origin of the cells in the efferent lymph from a single lymph node, in ‟Journal of experimental medicine", 1965, CXXI, pp. 901-911.
Harker, L. A., Megakaryocytes quantification, in ‟Journal of clinical investigation", 1968, XLVII, pp. 452-457.
Isaacs, R., Quantitative studies on the number of cells in the bone marrow, in ‟Journal of clinical investigation", 1932, II, p. 9.
Killman, S. A., Cronkite, E. P., Fliedner, T. M., Bond, V. P., Mitotic indices of human bone marrow cells. I. Number and distribution of mitoses, in ‟Blood", 1962, XIX, pp. 743-750.
Lajtha, L. G., Studies on the kinetics of erythropoiesis: a model of the erythron, in Ciba symposium on haemopoiesis (a cura di G. E. W. Wolstenholme e M. O'Connor), London 1960, pp. 289-314.
Leder, L. D., Der Blutmonocyt, Berlin-Heidelberg-New York 1967.
Ling, N. R., Lymphocyte stimulation, Amsterdam 1968.
Lo Bue, J. A., Gordon, A. S. e altri (a cura di), La dinamica della ematopoiesi, Roma 1968.
Lossen, J., Über das Verhalten des Knochenmarkes bei verschiedenen Erkrankungen des Kindesalters, in ‟Virchows Archiv für pathologische Anatomie und Physiologie und für klinische Medizin", 1910, CC, pp. 258-320.
Mc Culloch, E. A., Differentiation of hemopoietic stem cells, in Proceedings of XII congress of the International Society of Haematology, New York 1968, pp. 260-287.
Marchesi, V. T., Gowans, J. L., The migration of lymphocytes through the endothelium of venules in lymph nodes: an electron microscope study, in ‟Proceedings of the Royal Society" (Series B), 1963, CLIX, pp. 283-290.
Mechanik, N., Untersuchungen über das Gewicht des Knochenmarks des Menschen, in ‟Zeitschrift für Anatomie und Entwicklungsgeschichte", 1926, LXXIX, pp. 58-99.
Miller, J. F. A. P., Immunological function of the thymus, in ‟Lancet", 1961, II, pp. 748-749.
Miller, J. F. A. P., The thymus in relation to the development of immunological capacity, in Ciba symposium on the thymus: experimental and clinical studies (a cura di G. E. W. Wolstenholme e R. Porter), London 1966, pp. 153-174.
Miller, J. F. A. P., Mitchell, G. F., Thymus and antigen-reactive cells, in ‟Transplantation review", 1969, I, pp. 3-43.
Moore, M. A. S., Metcalf, D., Ontogeny of the haemopoietic system: yolk sac origin of in vivo and in vitro colony forming cells in the developing mouse embryo, in ‟British journal of haematology", 1970, XVIII, pp. 279-296.
Moore, M. A. S., Metcalf, D., Haemopoietic cells, Amsterdam-London 171.
Mori, Y., Lennert, K., Electron microscopic atlas of lymph node cytology and pathology, Berlin-Heidelberg-New York 1969.
Nossal, G. J. V., Ada, G. L., Antigens, lymphoid cells, and the immune response, New York 1971.
Nowell, P. C., Phytohaemagglutinin: initiator of mitosis in cultures of normal human leukocytes, in ‟Cancer research", 1960, XX, pp. 462-466.
Odell, T. T., Jackson, C. W., Gosslee, D. G., Maturation of rat megakaryocytes studied by microspectrophotometric measurement of DNA, in ‟Proceedings of the Society of Experimental Biology and Medicine", 1965, CXIX, pp. 1194-1199.
Pontoni, L., Su alcuni reperti citologici ricavati dal mielogramma, in ‟Haematologica", 1936, XVII, p. 883.
Roitt, I. M., Greavs, M. F., Torrigiani, G., Brostoff, J., Playfair, J. H. L., The cellular basis of immunological responses: a synthesis of some current views, in ‟Lancet", 1969, II, pp. 367-371.
Segerdahl, E., Über die Sternalpunktion, in ‟Acta medica scandinavica", 1935, LXIV, suppl., pp. 28-34.
Till, J. E., McCulloch, E. A., A direct measurement of the radiation sensitivity of normal mouse bone marrow cells, in ‟Radiation research", 1961, XIV, pp. 213-222.
Volkman, H., Gowans, J. L., The origin of macrophages from bone marrow, in ‟British journal of experimental pathology", 1965, XLVI, pp. 62-70.
Warner, N. L., Szenberg, A., The immunological function of the bursa of Fabricius, in the chicken, in ‟Annual review of microbiology", 1964, XVIII, pp. 253-268.
Weiss, L., The structure of the bone marrow: functional interrelationships of vascular and hemopoietic compartments in experimental hemolytic anemia. An electron microscopic study, in ‟Journal of morphology", 1965, CXVII, pp. 467-480.
Wetzel, G., Geschichtliches zur Bestimmung der Grösse des Markorgans, in ‟Zeitschrift für Zellforschung und Entwicklungsgeschichte", 1927, LXXXII, pp. 70-72.
Whang, J., Frei, E. III, Tjio, J. H., Carbone, P. P., Brecker, G., The distribution of the Philadelphia chromosome in patients with chronic myelogenous leukaemia, in ‟Blood", 1963, XXII, pp. 664-673.
Yoffey, J. M., Cellular equilibria in blood and blood forming tissues, in Homeostatic mechanisms, in ‟Brookhaven Symposia in Biology", 1957, X, pp. 1-30.
Anemie emolitiche
SOMMARIO: 1. Introduzione. □ 2. Eritropoiesi. □ 3. L'eritrocita. □ 4. Invecchiamento e distruzione dell'eritrocita. □ 5. Classificazione delle anemie. □ 6. Anemie ereditarie: a) anemie emolitiche causate da emoglobine anormali; b) anemie emolitiche provocate da una diminuita sintesi di emoglobina; c) difetti enzimatici dell'eritrocita; d) difetti della membrana eritrocitaria. □ 7. Anemie emolitiche acquisite: a) anemie emolitiche causate da agenti infettivi; b) anemie emolitiche provocate da agenti chimici o fisici; c) cause immunologiche di anemie emolitiche; d) anemie emolitiche secondarie a disordini endocrini o del sistema reticoloendoteliale. □ Bibliografia.
1. Introduzione.
L'eritrocita umano maturo ha una vita media di circa 120 giorni: ogni condizione che ne accorcia sostanzialmente la vita provoca un'anemia emolitica. Si conoscono varie cause di anemia emolitica: malattie congenite, malattie autoimmuni, agenti infettivi, ecc. Inoltre, possono essere incluse fra le anemie emolitiche quelle malattie in cui vi è distruzione delle cellule dalle quali derivano gli eritrociti.
Per poter trattare l'argomento nel modo più completo è necessario che la discussione sulle diverse forme di anemie sia preceduta da una breve descrizione dello sviluppo dell'eritrocita, del suo invecchiamento e della sua distruzione.
2. Eritropoiesi.
L'eritrocita umano maturo è una cellula libera e mobile, priva di nucleo, con un diametro di 7,5 μm e una forma discoidale biconcava; essa origina nel tessuto emopoietico da cellule genitrici nucleate ed entra normalmente in circolo dopo che il nucleo è stato espulso (v. sangue: Organi emopoietici).
Nel corso dello sviluppo dell'organismo da embrione ad adulto, il tessuto emopoietico si localizza in organi diversi. L'eritropoiesi embrionale, caratterizzata dalla presenza di grandi eritrociti nucleati che si formano nelle isole ematiche del sacco vitellino all'esterno dell'embrione propriamente detto, cessa approssimativamente dopo due mesi; a essa fa seguito l'eritropoiesi fetale, che comincia nel fegato e si svolge poi per un breve tempo anche nella milza e finalmente nel midollo osseo. Il normale tessuto emopoietico adulto è localizzato esclusivamente nel midollo osseo. L'eritrocita fetale è anucleato e ha la stessa forma e le stesse dimensioni dell'eritrocita adulto; da questo differisce però per molte importanti caratteristiche, quali il tipo dell'emoglobina, l'assenza di certi enzimi e alcune proprietà immunologiche della membrana cellulare. La localizzazione dell'eritropoiesi fetale nel midollo osseo comincia approssimativamente al quinto mese e diviene prevalente al nono mese: l'eritrocita prodotto alla fine della vita fetale ha proprietà in comune sia con gli eritrociti fetali, sia con quelli adulti. Le emoglobine fetale e adulta sono presenti insieme (come mostreremo) nello stesso eritrocita.
Una varietà di cellule del sangue e i loro precursori sono reperibili nel tessuto emopoietico del midollo osseo. Tali cellule sono raccolte in cordoni che giacciono fra i capillari sinusoidi decorrenti dalle arteriole periferiche verso la vena centrale longitudinale. Le cellule del sangue formate al di fuori dei capillari entrano in circolo passando nel loro intemo.
Tutte le cellule ematiche derivano da ceppi di cellule multipotenti e indifferenziate chiamate emocitoblasti, che si dividono attivamente per tutta la vita adulta (v. sangue: Organi emopoietici). Una frazione degli emocitoblasti si differenzia per dare origine alle cellule progenitrici dei diversi tipi di cellule sanguigne. Gli eritrociti si formano attraverso successivi eventi differenziativi che di norma hanno luogo simultaneamente alla divisione cellulare. La prima cellula riconoscibile come progenitore dell'entrocita è l'eritroblasto, caratterizzato da un grande nucleo e dall'assenza o da una quantità assai scarsa di emoglobina nel citoplasma. Successivamente l'eritroblasto si differenzia in normoblasto, in cui l'emoglobina è chiaramente presente. Quando i normoblasti si differenziano in reticolociti, la quantità di emoglobina nel citoplasma aumenta e il nucleo diventa progressivamente più piccolo e picnotico. Poiché la divisione cellulare ha luogo fino allo stadio di normoblasto, questo rappresenta la cellula nucleata della serie rossa che si osserva con maggiore frequenza nel midollo.
Il nucleo viene espulso dai normoblasti maturi che si differenziano in reticolociti. Queste cellule sono leggermente più grandi degli eritrociti, contengono mitocondri, ribosomi e tutti quei componenti cellulari che sono necessari per la sintesi proteica. Nei reticolociti prosegue la sintesi di emoglobina, che rappresenta il 95-98% delle proteine sintetizzate. Vengono sintetizzati anche gli enzimi necessari per il ruolo fisiologico dell'eritrocita, quali l'anidrasi carbonica e la metemoglobinareduttasi. I mitocondri e i ribosomi sono progressivamente distrutti da processi autolitici durante la maturazione del reticolocita, in un periodo inferiore ai due giorni. I reticolociti maturi entrano in circolo e si differenziano in eritrociti approssimativamente in 24-29 ore. L'intero processo differenziativo, da eritroblasto a eritrocita, ha luogo in una settimana circa. Nei pazienti con anemia emolitica questo processo può subire varie modificazioni. Il numero degli eritrociti e delle altre cellule ematiche in circolo è notevolmente costante in un ambiente stabile e in condizioni fisiologiche costanti. Variazioni nel numero degli eritrociti in circolo si verificano in seguito a modificazioni ambientali o fisiologiche che fanno aumentare o diminuire la domanda corporea di ossigeno per un periodo di tempo prolungato: per esempio, un aumento del numero di eritrociti si verifica tipicamente nel passaggio da bassa a elevata altitudine. L'aumento del numero di eritrociti viene ottenuto con la proliferazione nel midollo dei precursori delle cellule della serie rossa, che si risolve appunto in un'aumentata produzione di eritrociti. Stimoli diversi producono un aumento delle altre cellule ematiche circolanti; le infezioni, per esempio, determinano un aumento dei leucociti. Così la proliferazione dei precursori di ciascun tipo cellulare nel midollo osseo è regolata indipendentemente, mediante meccanismi precisi.
L'eritropoiesi è regolata dall'ormone eritropoietina, che è sintetizzato dalle cellule renali. L'eritropoietina è una glicoproteina che stimola la differenziazione delle cellule stipiti in eritroblasti e aumenta la velocità di sintesi dell'emoglobina negli eritroblasti e nei normoblasti. L'ipossia stimola la sintesi di eritropoietina; quando la capacità di trasporto di ossigeno del sangue è diminuita, come nelle anemie, la sintesi di eritropoietina viene stimolata e la produzione di eritrociti viene conseguentemente aumentata.
La produzione delle altre cellule del sangue è regolata, indipendentemente, da altri ormoni, dei quali soltanto alcuni sono stati identificati.
3. L'eritrocita.
L'eritrocita maturo è sostanzialmente caratterizzato dal fatto di contenere una soluzione altamente concentrata di emoglobina all'interno di una membrana relativamente elastica: in esso mancano gli organuli intracellulari e l'emoglobina, non interamente in soluzione dato che le sue molecole sono disposte in un reticolo semiordinato nel citoplasma viscoso, rappresenta circa il 97% di tutte le proteine (v. sangue: Emoglobina).
Poiché l'emoglobina è il componente principale degli eritrociti, dal suo comportamento fisico-chimico dipende la maggior parte delle caratteristiche fisiologiche di tali elementi; la molecola di emoglobina - un'emoproteina, costituita da una parte proteica (globina) e da un gruppo prostetico (eme) - è formata da due paia di catene identiche così che in ciascuna delle quattro catene polipeptidiche è presente un gruppo eme. L'eme è una porfirina (protoporfirina IX) che contiene un atomo di ferro bivalente, il quale può legarsi con una molecola di ossigeno.
I due tipi di catene polipeptidiche dell'emoglobina umana adulta (Hb A) sono stati chiamati catene α e β, e sono formati rispettivamente da 141 e 146 amminoacidi. Le catene α sono presenti anche nell'emoglobina fetale (Hb F) e in quella componente minore dell'emoglobina dell'adulto chiamata emoglobina A2 (Hb A2). Queste emoglobine differiscono dall'Hb A per quanto riguarda le altre catene polipeptidiche: sono infatti presenti catene γ nell'Hb F e catene δ nell'Hb A2, che hanno sequenza di amminoacidi diversa, ma struttura tridimensionale quasi identica a quella della catena β. In una molecola di emoglobina le quattro catene polipeptidiche sono disposte in modo tale che due piani tra loro perpendicolari dividono la molecola in due metà identiche (v. fig. 1).
Le molecole dell'emoglobina si dissociano reversibilmente in due mezze molecole identiche:
(emoglobina adulta) α2β2⇄2αβ (mezze molecole).
Questa dissociazione ha luogo spontaneamente in soluzioni diluite di emoglobina, ma non è ancora chiaro se essa abbia un ruolo significativo nell'attività fisiologica dell'emoglobina. Una molecola di emoglobina può legare quattro molecole di ossigeno - una per ciascuna catena peptidica - senza che gli atomi di ferro dell'eme vengano ossidati. La liberazione dell'ossigeno determina variazioni nella dimensione molecolare dell'atomo di ferro, che protrude dal piano dell'eme (l'atomo di ferro occupa la parte centrale dell'anello protoporfirinico), e provoca una complessa serie di alterazioni strutturali all'interno di ciascuna catena polipeptidica.
La curva di dissociazione dell'ossiemoglobina ha un caratteristico andamento sigmoidale; questo indica che il legame della prima molecola di ossigeno facilita quello delle successive, aumentando l'affinità dell'emoglobina per l'ossigeno (v. fig. 2). Tale fenomeno è alla base dell'attività fisiologica dell'emoglobina come vettore di ossigeno: l'emoglobina, infatti, si combina rapidamente con l'ossigeno nei polmoni, dove la concentrazione relativa di ossigeno è alta, e lo cede invece ai tessuti, dove tale concentrazione è bassa. La liberazione dell'ossigeno dall'emoglobina è favorita anche dall'abbassamento di pH provocato dalla liberazione di CO2 dai tessuti (effetto Bohr). Nel momento in cui la forma ossigenata dell'emoglobina (ossiemoglobina) si trasforma nella forma non ossigenata (desossiemoglobina), alcuni gruppi ionizzati negativamente, in seguito a complessi cambiamenti strutturali delle catene polipeptidiche dell'emoglobina, divengono accessibili e si combinano con protoni (H+), esercitando quindi un importante effetto ‛tampone' nel mantenere costante il pH del sangue.
Il ruolo fisiologico dell'eritrocita è così altamente specializzato nel trasporto dell'ossigeno e del CO2 che le sue proprietà si sono evolute specialmente per facilitare questa funzione. Cosi, l'eritrocita necessita di relativamente poca energia per sopravvivere e questa energia non deriva dall'utilizzazione del glucosio nel ciclo di Krebs, ma dalla glicolisi nella via metabolica di Embden-Meyerhof e in quella del pentoso-fosfato. Nella glicolisi anaerobica il glucosio è metabolizzato a lattato, mentre nella via metabolica del pentoso-fosfato il glucosio è ossidato a pentoso-monofosfato e metabolizzato successivamente in altri derivati fosforilati. Solo il 10% circa del glucosio è metabolizzato dall'entrocita attraverso questa seconda via metabolica.
Oltre agli enzimi necessari per il metabolismo del glucosio, altri enzimi sono presenti in quantità relativamente abbondante negli eritrociti: la catalasi, che scinde l'H2O2 formatasi nel globulo rosso; l'anidrasi carbonica, che catalizza la formazione e la dissociazione di carbonato da e in CO2; la metemoglobinareduttasi, che riduce l'emoglobina contenente ferro trivalente (metemoglobina); la purin-nucleotidefosforilasi; la colinesterasi; la fosfatasi acida; la glutammico-ossalacetico-transaminasi.
Particolarmente importante per il suo effetto sulla funzione dell'emoglobina è il 2,3-difosfoglicerato. Questo composto è formato, a opera dell'enzima difosfogliceratomutasi, dall'1,3-difosfoglicerato, che è uno dei metaboliti del glucosio nella glicolisi anaerobica. La maggior parte del fosfato organico presente negli eritrociti è sotto forma di 2,3-difosfoglicerato. Una molecola di questo composto si combina con una molecola di emoglobina riducendone l'affinità per l'ossigeno, senza cambiare la forma sigmoide della curva di ossigenazione. Lo si può quindi considerare come un cofattore che facilita la liberazione dell'ossigeno dall'emoglobina.
Gli eritrociti sono molto stabili in soluzioni isotoniche, donde l'uso corrente di conservare il sangue per trasfusione fino a 21 giorni a O °C. Tuttavia gli eritrociti in soluzioni non isotoniche vanno rapidamente incontro alla lisi. Il fenomeno della lisi è dovuto alla proprietà della membrana di queste cellule di essere liberamente permeabile all'acqua e ai piccoli anioni come il cloruro e il bicarbonato. La rapida penetrazione dell'acqua negli eritrociti in soluzioni ipotoniche, causata dalla differenza di pressione osmotica, ne provoca il rigonfiamento e la lisi.
Le caratteristiche della membrana eritrocitaria risultano essenziali per l'attività fisiologica dell'eritrocita e consentono inoltre molti fenomeni di identificazione immunologica. Le proprietà fisiologiche peculiari di tale membrana sono importanti per il passaggio, mediante diffusione o trasporto attivo, di sali e di metaboliti nel citoplasma: il glucosio, ad esempio, è trasportato attivamente, probabilmente per mezzo di una permeasi specifica. Nella membrana sono anche presenti gli enzimi che idrolizzano l'ATP (ATPasi) in concomitanza con il trasporto di cationi: l'idrolisi di ATP e quindi processi metabolici che producono energia sono necessari per mantenere all'interno degli eritrociti un'alta concentrazione di K+ e una bassa concentrazione di Na+, in contrasto con la bassa concentrazione di K+ e l'alta concentrazione di Na+ esistente nel plasma. Sulla membrana, poi, sono presenti diversi lipopolisaccaridi, la cui struttura è sotto controllo genetico e costituisce la base chimica per la determinazione del gruppo sanguigno; tali lipopolisaccaridi sono i recettori per gli anticorpi che agglutinano gli eritrociti o causano la loro lisi.
4. Invecchiamento e distruzione dell'eritrocita.
Gli eritrociti hanno una vita media di circa 120 giorni, dopodiché vengono eliminati; la loro distruzione avviene in seguito al normale processo di invecchiamento o a causa di anomalie cellulari. L'invecchiamento è una conseguenza della perdita di attività degli enzimi, che non possono essere rimpiazzati dato che gli eritrociti non sintetizzano alcuna proteina.
Una diminuzione delle attività enzimatiche è dimostrata dal fatto che gli eritrociti vecchi hanno un'attività metabolica ridotta: infatti, diminuisce la glicolisi e divengono meno attivi anche altri processi che sono importanti per la funzione eritrocitaria, come il trasporto del sodio e del potassio. La distruzione degli eritrociti invecchiati avviene con tutta probabilità a seguito di una loro frammentazione; i frammenti, ancora avvolti dalla membrana cellulare così che pochissima emoglobina viene rilasciata libera nel circolo, sono poi trattenuti dal sistema reticoloendoteliale, principalmente nella milza, e quindi digeriti dalle cellule fagocitarie.
La scarsa quantità di emoglobina che passa in circolo si combina con alta affinità con l'aptoglobina, una proteina che ha la funzione specializzata di rimuovere l'emoglobina dal plasma. La globina, la parte proteica dell'emoglobina, è metabolizzata in amminoacidi, che possono poi venire riutilizzati nella biosintesi delle proteine; l'eme viene degradato mediante rimozione dell'atomo di ferro e apertura dell'anello porfirinico, attraverso alcune tappe metaboliche intermedie. Si forma un metabolita colorato, chiamato bilirubina, che viene trasportato dall'albumina alle cellule epatiche: queste cellule convertono la bilirubina, assai scarsamente solubile in acqua, in un suo derivato diglucuronidico idrosolubile, che viene quindi escreto con la bile e poi trasformato dai batteri del grosso intestino, attraverso una serie di riduzioni enzimatiche, in urobilinogeno.
5. Classificazione delle anemie.
Sono state proposte diverse classificazioni delle anemie emolitiche, basate ora sulla loro eziologia, ora sulla natura del processo fisiologico alterato, sul quadro clinico, o ancora sulle caratteristiche morfologiche degli eritrociti e delle cellule del midollo osseo. Per una descrizione razionale di questo gruppo di malattie, la classificazione più soddisfacente è quella basata sull'eziologia di ciascuna forma di anemia emolitica: sfortunatamente, poiché l'eziologia di alcune di esse non è ancora conosciuta, ne è impossibile una classificazione corretta. Comunque, dal momento che è abbastanza ben conosciuta l'eziologia della grande maggioranza delle anemie, verrà qui adottata una classificazione eziologica (v. tabella).
Un primo gruppo di anemie è causato dalla trasmissione di geni anormali. Queste anomalie genetiche sono piuttosto frequenti in alcune popolazioni (v. cap. 6) e possono interessare ciascun componente dell'eritrocita: emoglobina, enzimi, membrana eritrocitaria. Le manifestazioni di geni anormali a livello molecolare consistono in una sostituzione di uno o più amminoacidi nella sequenza di una catena polipeptidica, oppure in una riduzione della velocità di sintesi di una catena polipeptidica. Molto spesso un gene anormale può determinare ambedue gli effetti.
Un cambiamento di amminoacidi nella sequenza può provocare un'alterazione notevole nelle caratteristiche fisico-chimiche di una molecola proteica, causando o un cambiamento delle sue proprietà catalitiche, o una relativa instabilità. La modificazione della velocità di sintesi può diminuire la concentrazione cellulare di una proteina, portandola a un livello spesso insufficiente per lo svolgimento della sua attività fisiologica.
Sia un cambiamento nelle proprietà fisico-chimiche dell'emoglobina, sia una riduzione della sua velocità di sintesi sono causa di distruzione prematura dell'eritrocita.
Anche un difetto di uno degli enzimi che sono essenziali per il metabolismo dell'eritrocita causa il suo prematuro invecchiamento e la sua distruzione. La distruzione degli eritrociti che possiedono una quantità di emoglobina minore del normale è probabilmente causata dall'aumentata velocità di frammentazione ed emolisi di tali cellule, che sono relativamente piccole (microciti). La distruzione degli eritrociti contenenti un'emoglobina anormale può essere causata da un cambiamento dello stato fisico dell'emoglobina al loro interno, durante i fenomeni di aggregazione o di precipitazione: questi fenomeni diminuiscono l'elasticità dell'eritrocita e interferiscono con il suo metabolismo, affrettandone l'invecchiamento e la distruzione.
Un altro gruppo di anemie è provocato da condizioni acquisite: deficienze nutrizionali, agenti infettivi, malattie immunologiche e agenti chimici o fisici.
Un gruppo di anemie che possono essere considerate acquisite è quello che comprende le anemie secondarie a disturbi endocrini o a malattie del sistema reticoloendoteliale.
Le anemie a eziologia nutrizionale possono essere causate da deficienze di ciascuno degli elementi che sono necessari per la formazione dell'eritrocita. In particolare, una deficienza di ferro diminuisce la velocità di sintesi dell'eme e quindi anche quella dell'emoglobina. Ma in generale qualsiasi deficienza nutritiva ha un effetto profondo e immediato sul tempo di formazione degli eritrociti, poiché questi sono tra le cellule che proliferano costantemente e più attivamente.
6. Anemie ereditarie.
Le anemie ereditarie sono relativamente comuni in molte popolazioni che sono state esposte alla malaria per secoli. Le forme più diffuse sono quelle che producono un cambiamento della struttura dell'emoglobina o degli enzimi eritrocitari, oppure una diminuzione della velocità di sintesi dell'emoglobina. Queste anomalie sono presenti dalla nascita, hanno un considerevole effetto sul metabolismo degli eritrociti e ne accorciano notevolmente la sopravvivenza. È questa relativa fragilità degli eritrociti che conferisce una certa protezione nei confronti dei parassiti malarici. Gli eritrociti vengono invasi dai parassiti (v. cap. 7, È a), i quali si sviluppano nel loro interno e vengono poi rilasciati in circolo quando maturano, dopo un intervallo di tempo caratteristico per ciascun tipo di parassita.
La relativa resistenza verso i parassiti malarici di individui portatori di un difetto eritrocitario sembra dovuta al fatto che in questi soggetti la lisi eritrocitaria ha luogo molto facilmente: è probabile che una lisi prematura dell'eritrocita non permetta la maturazione dei parassiti, che sono quindi rilasciati in circolo immaturi e incapaci di infestare altri eritrociti.
Alcuni difetti congeniti della struttura dell'emoglobina o una forte riduzione della sua velocità di sintesi hanno relativamente poca importanza negli eterozigoti, mentre provocano una forte anemia negli omozigoti: questi individui spesso non vivono oltre la pubertà e non raggiungono quindi l'età riproduttiva. Malgrado questa perdita di omozigoti e quindi di copie dei geni di emoglobina anormale, la frequenza relativa di tali geni è rimasta abbastanza alta in molte popolazioni, il che indica che il vantaggio selettivo di cui godono gli eterozigoti è sufficiente a compensare la perdita degli omozigoti. Questi divengono una frazione importante della progenie in una popolazione in cui la frequenza di un gene anormale è alta (la frequenza degli omozigoti affetti nella progenie è uguale a 1/4 del quadrato della frequenza degli eterozigoti nella generazione parentale; v. eugenica).
Molti studi hanno mostrato che il vantaggio selettivo degli eterozigoti è notevole in aree dove la malaria è endemica; donne eterozigoti hanno più figli di donne normali, e anche se alcuni di questi bambini sono omozigoti e muoiono durante la fanciullezza, il numero dei discendenti è tale da compensare la loro perdita. È quindi facilmente comprensibile come sia possibile raggiungere e mantenere una frequenza relativamente elevata di geni che provocano anomalie dell'emoglobina nel caso che la malaria rimanga un fattore selettivo importante. Con l'eradicazione della malaria da diverse aree del mondo, la frequenza dei geni di emoglobine anormali subirà un decremento proporzionale alla frequenza con la quale sono generati omozigoti malati.
Quindi la perdita di geni di emoglobina anormale sarà piuttosto alta in popolazioni con alta frequenza di tali geni, divenendo sempre più bassa col diminuire della loro frequenza. Pertanto, anche quando la malaria sarà completamente scomparsa e i geni per le emoglobine anormali non offriranno più alcun vantaggio selettivo, le anemie emolitiche ereditarie saranno ancora relativamente frequenti.
a) Anemie emolitiche causate da emoglobine anormali.
La presenza di un'emoglobina anormale può causare anemie emolitiche in molti modi diversi. Finora nell'uomo sono stati scoperti più di un centinaio di tipi di emoglobine; queste per la maggior parte sono state riscontrate in modo occasionale nel corso di indagini su varie popolazioni e determinano un modesto effetto patologico sui loro portatori, o ne sono affatto prive. Alcune di queste emoglobine anormali, a causa della loro relativa rarità, non sono quasi mai osservate nella condizione di omozigosi e gli eterozigoti non accusano alcun sintomo.
Altri tipi, invece, sono abbastanza frequenti e causano ben delineate anemie negli omozigoti e, in qualche caso, anche negli eterozigoti.
Anemia a cellule falciformi (drepanocitosi). - Questa anemia emolitica è causata da un'emoglobina anormale, chiamata emoglobina S, che nello stato non ossigenato è scarsamente solubile, così che le sue molecole si riuniscono in grossi aggregati semicristallini in grado di deformare gli eritrociti: in tal modo si determina la comparsa di cellule a forma di falce, caratteristicamente presenti nel sangue venoso (v. fig. 3). Questo comportamento anormale dell'emoglobina S è provocato dalla sostituzione di un singolo amminoacido: un residuo valinico nella sesta posizione a partire dall'N-terminale della catena β sostituisce nell'emoglobina S un residuo glutammico; la presenza del residuo valinico in questa posizione causa la formazione di una struttura ad anello e l'aggregazione di molecole di emoglobina in larghe supereliche.
Gli eritrociti falciformi sono più rigidi di quelli normali e non passano facilmente attraverso i piccoli capillari. Eritrociti di 7,5 μm di diametro possono passare normalmente attraverso capillari di diametro minore (fino a 3 μm) a causa della loro elasticità, mentre gli eritrociti falciformi ostruiscono i piccoli capillari e causano trombosi.
Solo gli omozigoti sono gravemente colpiti, mentre gli eterozigoti hanno scarsi sintomi o ne sono del tutto privi. Gli individui affetti presentano anemia cronica, dovuta alla lisi e alla distruzione prematura degli eritrociti falciformi, ittero, dolori addominali e articolari. L'ittero è un sintomo molto comune di tutte le anemie emolitiche gravi ed è provocato dall'aumentato metabolismo dell'eme e dei suoi derivati. I dolori addominali e articolari sono invece tipici della falcemia, e spesso si manifestano come attacchi improvvisi, accompagnandosi a progressivo senso di debolezza e affaticamento. A carico delle articolazioni si possono osservare dolorabilità e tumefazione. Vi può essere un lieve aumento di volume della milza, ma una splenomegalia rilevabile alla palpazione è presente solo in pochi pazienti. Il cuore è in genere ingrossato e le aritmie sono frequenti.
Nei primi anni di vita le mani e i piedi possono essere tumefatti e dolenti. Nei casi gravi i pazienti hanno un aspetto fisico caratteristico, con mani e piedi lunghi e stretti e con un tronco piuttosto corto. Le ulcere croniche delle gambe sono frequenti sia negli adolescenti sia negli adulti. Deformità ossee, quali quelle del cranio, possono determinarsi in conseguenza dell'intensa eritropoiesi.
L'anemia a cellule falciformi, descritta dapprima nei Negri del Nordamerica, è molto frequente in Africa, soprattutto nelle regioni centrali e orientali. La sua incidenza varia considerevolmente da tribù a tribù ed è correlata con quella della malaria: non è affatto raro trovare tribù con una frequenza di eterozigoti del 30-40%. Al di fuori dell'Africa l'anemia a cellule falciformi è stata osservata in alcuni paesi mediterranei in individui di origine greca o siciliana, nell'Arabia meridionale, nei negroidi Veddidi dell'India meridionale e nella Turchia meridionale.
La diagnosi di anemia a cellule falciformi è basata sul reperto di eritrociti falciformi e sull'analisi elettroforetica dell'emoglobina. La formazione di eritrociti falciformi può essere osservata quando una goccia di sangue viene tenuta fuori del contatto dell'aria sotto un vetrino coprioggetti, in modo da provocare la deossigenazione dell'emoglobina, oppure aggiungendo al sangue un agente riducente, come sodio metabisolfito (disolfito) al 2%, che accelera la deossigenazione dell'emoglobina. L'analisi elettroforetica è importante per stabilire la possibile presenza di altre emoglobine anormali; la formazione di eritrociti falciformi ha luogo quando l'emoglobina S è presente assieme all'emoglobina C o ad altre emoglobine anormali rare.
Finora non è stato scoperto alcun trattamento specifico dell'anemia a cellule falciformi. Farmaci vasodilatatori sono usati per il trattamento delle crisi dolorose. Recenti pubblicazioni che segnalavano l'uso di agenti che riducono la formazione di legami idrogeno, come l'urea, in grado quindi di prevenire l'aggregazione delle molecole di emoglobina, devono ancora ricevere la necessaria conferma clinica.
Anemia da emoglobina C. - Si tratta di una rara anemia ereditaria provocata da una condizione di omozigosi per l'emoglobina C, un'emoglobina anormale nella quale lo stesso residuo amminoacidico che nell'emoglobina 5 viene sostituito da un residuo valinico viene invece sostituito da una lisina. L'emoglobina C è stata descritta tipicamente nei Negri e la sua massima incidenza è stata riscontrata nel Ghana settentrionale. Casi di pazienti con emoglobina C sono stati inoltre osservati in Algeria e in Sicilia.
I pazienti affetti da malattia da emoglobina C presentano una modesta anemia e splenomegalia e, con minor frequenza, accusano dolori addominali senza precisa localizzazione: la maggior parte di questi sintomi dipende dalla diminuzione della vita media degli eritrociti (che scende a 30-55 giorni), caratteristica di tale condizione. Gli omozigoti affetti vivono bene e molti pazienti sono vissuti per un tempo normale.
Malattia da emoglobina M. - Le emoglobine anormali chiamate emoglobina M sono caratterizzate dalla tendenza a formare facilmente metemoglobina in normali condizioni fisiologiche: esse non determinano anemia emolitica e sono qui descritte solo per fornire un quadro più completo degli effetti di emoglobine anormali.
Ciascun tipo di emoglobina M corrisponde a una diversa sostituzione di un amminoacido nelle catene polipeptidiche dell'emoglobina: tali sostituzioni interessano residui di amminoacidi che interagiscono con l'eme, per cui una loro alterazione interferisce con la riduzione dell'atomo di ferro allo stato ferroso. Nell'emoglobina M l'atomo di ferro della catena peptidica in cui è presente la sostituzione è ossidato permanentemente alla forma ferrica. In tale condizione la capacità di trasporto dell'ossigeno degli eritrociti è ridotta e gli individui affetti (sono stati osservati solo eterozigoti) mostrano cianosi, provocata dal caratteristico colore della metemoglobina, e possono accusare debolezza e affaticamento; la prognosi, tuttavia, è buona.
Malattia da emoglobina instabile. - Un gruppo di anemie emolitiche è provocato dall'ereditarietà di emoglobine anormali caratterizzate da notevole instabilità; tali emoglobine precipitano all'interno degli eritrociti, formandovi corpi di inclusione identificabili all'osservazione microscopica e conosciuti come corpi di Heinz.
La presenza di un'emoglobina anormale instabile, tuttavia, può essere dimostrata soltanto scaldando soluzioni di emoglobina a 50 o a 70 °C per 1 ora: si può così formare un precipitato visibile, mentre l'analisi elettroforetica della soluzione di emoglobina eseguita prima e dopo riscaldamento consente di mettere in evidenza la scomparsa di un componente instabile.
Le emoglobine instabili sono piuttosto rare. È stato dimostrato che esse si formano in seguito a varie sostituzioni amminoacidiche (sia nella catena a che nella catena β), ognuna delle quali determina condizioni in contrasto con quelle necessarie a formare una molecola proteica stabile: per esempio, un residuo polare ne sostituisce uno non polare, oppure una prolina sostituisce un altro amminoacido in una regione a elica di una catena peptidica, interferendo quindi con il suo ripiegamento corretto.
Tali emoglobine sono state riscontrate solo in eterozigoti e si ritiene che la condizione di omozigosi sia letale. La prognosi è relativamente favorevole; in alcuni casi l'asportazione della milza migliora la sopravvivenza degli eritrociti.
Instabilità di emoglobine anormali indotta da farmaci. - Si sono segnalati limitati casi di famiglie, alcuni membri delle quali sono risultati portatori di un'emoglobina anormale che diventa marcatamente instabile dopo somministrazione di certi farmaci. Un esempio tipico è l'emoglobina Zurigo, che precipita formando corpi di inclusione dopo somministrazione di solfammidici od ossichinoloni. La somministrazione per via orale di questi farmaci può provocare gravi crisi emolitiche nei portatori di queste emoglobine anormali.
b) Anemie emolitiche provocate da una diminuita sintesi di emoglobina.
Sono classificate come talassemie alcune anemie ereditarie caratterizzate da una diminuzione della velocità di sintesi dell'emoglobina. La prima chiara descrizione di queste forme morbose fu data da Cooley e Lee nel 1925, e pertanto si convenne di designarle con la denominazione generica di anemie di Cooley. Tuttavia, poiché in base alla loro eziologia possono essere differenziate forme diverse, è successivamente sembrato più appropriato usare il termine ‛talassemie' derivato dal vocabolo greco ϑάλασσα (mare): queste anemie sono state infatti osservate con alta incidenza, anche se non esclusivamente, nelle popolazioni mediterranee di origine italiana e greca. Le prime osservazioni di Cooley furono eseguite su pazienti provenienti da regioni mediterranee, ma l'ereditarietà di questa anemia non è stata chiarita fino al 1940.
In seguito è stato accertato che in forme diverse di talassemia è inibita o abolita la sintesi di una delle catene peptidiche dell'emoglobina. L'attuale classificazione delle talassemie si basa appunto su questa osservazione; così le α-talassemie sono quelle forme in cui è ridotta la sintesi delle catene α, le β-talassemie quelle in cui è ridotta la sintesi delle catene β. Vi sono altre forme di talassemie, nelle quali manca completamente la sintesi delle due catene β e δ, che sono state chiamate βδ-talassemia e anemia Lepore; queste sono provocate da alterazioni funzionali dei geni β e δ consistenti, rispettivamente, in una loro soppressione completa o parziale, tale che i due geni risultano fusi assieme, dando luogo alla formazione della cosiddetta ‛catena Lepore'. I meccanismi molecolari che provocano la diminuita sintesi di emoglobina nelle α e β-talassemie non sono conosciuti.
La diminuzione della velocità di sintesi di una delle catene dell'emoglobina può essere modesta o vi può essere una soppressione completa della sintesi di un tipo di catena peptidica: conseguentemente forme diverse di talassemia possono mostrare diversa gravità, in relazione alla quantità di emoglobina che il paziente è in grado di sintetizzare. L'emoglobina presente nei pazienti talassemici omozigoti è normale nella struttura e nella sequenza di amminoacidi.
I geni talassemici sono uniti ai geni strutturali per le corrispondenti catene emoglobiniche (non sono uniti i geni per le catene α e β). Questo fatto ha suggerito l'ipotesi che la mutazione talassemica possa alterare qualche elemento regolatore che controlla la velocità di sintesi dell'emoglobina.
La forma omozigote della talassemia, la cosiddetta thalassemia major, è una malattia letale che di norma non consente la sopravvivenza oltre l'adolescenza degli individui che ne sono affetti, anche se forniti della migliore assistenza medica. Della forma eterozigote si distinguono un tipo completamente asintomatico, denominato thalassemia minima, e altri caratterizzati da disturbi di varia gravità e prognosi più o meno buona, cui si dà il nome rispettivamente di thalassemia minor e di thalassemia intermedia. Gli eritrociti dei talassemici eterozigoti sono più piccoli di quelli normali, dato che contengono una minore quantità di emoglobina, sono cioè microciti, donde il termine di microcitemia con il quale viene anche designata la talassemia.
La caratteristica più saliente della thalassemia major consiste nella notevole proliferazione del midollo, che è causa di alterazioni ossee, specialmente a carico del cranio; altra caratteristica è l'addome prominente in conseguenza della splenomegalia e spesso anche della epatomegalia. Nel cranio vi è di solito un notevole ispessimento della diploe e le radiografie mostrano una caratteristica struttura a spazzola dipendente dall'iperostosi di tipo spiculare reattiva all'iperplasia midollare. Le ossa lunghe appaiono ingrossate. Gli eritrociti presentano una morfologia caratteristica: molto eterogenei per quanto riguarda le dimensioni, hanno una forma distorta e contengono poca emoglobina. In circolo possono essere presenti cellule eritroidi nucleate (normoblasti). Gli eritrociti hanno un'esistenza brevissima e sono rapidamente distrutti nella milza, che diventa iperplasica; il loro numero è notevolmente ridotto e scende molto al di sotto del livello normale.
La grave anemia stimola il sistema eritropoietico a produrre una maggiore quantità di cellule, ma l'incapacità di produrre una quantità sufficiente di emoglobina rende queste cellule fortemente anormali ed è causa di un'eritropoiesi inefficiente. La stimolazione cronica del sistema eritropoietico provoca la formazione di cloni di cellule eritroidi che sintetizzano emoglobina fetale. Gli eritrociti che contengono emoglobina fetale sopravvivono più a lungo e molti dei pazienti affetti da thalassemia major hanno in circolo quasi esclusivamente emoglobina fetale. La sintesi di questa emoglobina, che compensa parzialmente la mancata sintesi di emoglobina adulta, ha luogo solo quando il sistema eritropoietico ha proliferato oltre la normalità, e ha quindi scarso effetto nel migliorare le condizioni generali dei malati.
È interessante rilevare che sono stati scoperti individui clinicamente normali sotto ogni aspetto che possiedono solo emoglobina fetale per tutta la vita; questi individui sono portatori di un gene mutante, che provoca la persistenza ereditaria dell'emoglobina fetale. Se si trovasse il modo di indurre i malati di talassemia a sintetizzare emoglobina fetale, piuttosto che tentare invano la sintesi di emoglobina adulta, si riuscirebbe forse a salvarli. Sfortunatamente, il meccanismo che controlla il passaggio dalla sintesi di emoglobina fetale a quella di emoglobina adulta non è ancora stato individuato.
α-talassemia. - La catena α è presente sia nell'emoglobina fetale sia nell'emoglobina adulta. L'incapacità di sintetizzare una quantità sufficiente di catene α, e quindi di emoglobina, è incompatibile con la sopravvivenza del feto: gli omozigoti per forme gravi di α-talassemia muoiono quindi in utero, mentre i soli individui affetti che sopravvivono sono quelli nei quali la sintesi della catena α è ridotta, ma non soppressa.
L'α-talassemia è più frequente nei paesi dell'Asia orientale che in quelli mediterranei. La maggior parte dei casi di morte intrauterina e di grave idrope fetale dovuti ad atalassemia omozigote sono stati osservati in Malesia e in Tailandia.
Il quadro clinico dell'α-thalassemia major è simile a quello della β-talassemia, con la differenza che la gravità della malattia varia notevolmente. Gli individui affetti sono spesso eterozigoti per due diversi geni α-talassemici, uno solo dei quali sopprime o diminuisce notevolmente la sintesi della catena α. In essi è presente un quadro emoglobinico caratteristico: poiché le catene β e γ sono prodotte in quantità normale, vi è un grande eccesso di queste molecole, che si aggregano e formano tetrameri β4 e γ4 (nel caso in cui vi sia sintesi compensatoria di emoglobina fetale). Queste molecole emoglobiniche sono altamente instabili e hanno una forte affinità per l'ossigeno, tale da renderle inutili ai fini del trasporto e della cessione di ossigeno ai tessuti. Nel sangue periferico di malati di α-talassemia è frequentemente possibile osservare nell'interno degli eritrociti la presenza di corpi di inclusione, la cui formazione può essere indotta in vitro trattando campioni di sangue con agenti che provocano la precipitazione delle instabili emoglobine tetrameriche.
β-talassemia. - La β-talassemia è la forma più frequente di anemia ereditaria nei paesi mediterranei. È stato stimato che i portatori di geni talassemici in Italia, per esempio, sono circa il 2% della popolazione. L'alta frequenza di questi geni dovrebbe far sì che vi sia 1 neonato talassemico ogni 10.000 nati, assumendo come del tutto casuale la probabilità che due talassemici si sposino. Tuttavia, la frequenza della talassemia è molto variabile e raggiunge valori estremamente alti in regioni dove la malaria è stata endemica, quali, in Italia, il delta del Po, la Sicilia e la Sardegna. In queste regioni la talassemia è una delle cause più comuni di mortalità durante l'adolescenza.
Il quadro clinico della thalassemia major è stato già descritto. Il quadro emoglobinico è abbastanza variabile: la normale emoglobina A adulta è presente assieme a una quantità variabile di emoglobina fetale (dal 20 al 90%). L'emoglobina A2 è caratteristicamente aumentata, sia in quantità assoluta sia relativamente all'emoglobina A. Anche negli eterozigoti si riscontra un aumento del livello dell'emoglobina A2 (thalassemia intermedia, minor e minima) che raggiunge valori anche doppi del normale; questo aumento rappresenta il criterio diagnostico più preciso per l'individuazione di eterozigoti per β-talassemia.
Nessuna terapia è disponibile oggi per curare le talassemie, eccettuate trasfusioni periodiche di sangue, che permettono di prolungare l'esistenza degli individui colpiti, e la splenectomia per migliorare la sopravvivenza delle cellule trasfuse. La sola misura atta a prevenire la thalassemia major è l'individuazione degli eterozigoti, che dovrebbero essere scoraggiati dallo sposarsi tra loro: quando due eterozigoti sono sposati, dovrebbero essere informati che la probabilità di avere un figlio affetto da thalassemia major è molto alta.
βδ-talassemia e anemia Lepore. - Queste anomalie ereditarie sono meno frequenti delle talassemie α e β, ma sono presenti nelle stesse popolazioni. Probabilmente anche queste anemie conferiscono una qualche protezione verso la malaria, ma non hanno raggiunto la stessa diffusione delle altre forme di talassemia, forse perché la protezione è meno efficace. Sono stati osservati solo pochissimi individui omozigoti, ma vi sono molti individui eterozigoti per una di queste anomalie e per la β-talassemia. Questi malati mostrano un quadro clinico simile a quello della thalassemia major. Generalmente gli omozigoti per la βδ-talassemia o per l'anemia Lepore mostrano un quadro clinico meno grave di quello della thalassemia major.
L'anemia Lepore è stata così chiamata dal nome di un malato di origine italiana, nel quale fu individuata l'anormale emoglobina tipica di questa forma di anemia: questa è formata da catene α normali, mentre le altre catene, chiamate catene Lepore, sono simili in parte alle β e in parte alle δ. La sequenza di queste catene riflette l'evento genetico che ha causato la delezione di parte dei geni β e δ. Questa delezione è probabilmente provocata da un irregolare crossing-over, avvenuto in un antenato dei portatori del gene Lepore, che viene trasmesso come gene semidominante mendeliano (v. eredità biologica; v. genetica: Citogenetica).
Drepanocitosi-β-talassemia. - Nel 1946 E. Silvestroni e I. Bianco descrissero una particolare forma di anemia emolitica caratterizzata dalla presenza nello stesso individuo delle due anomalie eritrocitarie drepanocitosi e microcitosi. Accurati studi hanno consentito di dimostrare che la malattia, cui fu dato il nome di Silvestroni-Bianco, è riscontrabile nelle popolazioni dell'Africa settentrionale, in quelle mediterranee di origine greca e italiana (specialmente siciliane e calabresi) e fra la popolazione negra dell'America, ed è causata dalla contemporanea presenza del gene per l'emoglobina S e di quello per la β-talassemia: in tale condizione, poiché la normale emoglobinogenesi è notevolmente ostacolata da quest'ultimo gene, quello per la drepanocitosi acquista una maggiore espressività. Ne consegue un contenuto eritrocitario di emoglobine anormali, rappresentate prevalentemente da Hb S, che può arrivare fino al 70%, e da circa il 25% di Hb F e A2, responsabile di un quadro clinico sovrapponibile a quello della drepanocitosi. La terapia della malattia è puramente sintomatica e la splenectomia si è dimostrata inefficace.
c) Difetti enzimatici dell'eritrocita.
Il metabolismo degli eritrociti è totalmente dipendente dall'utilizzazione del glucosio. L'energia generata dalla glicolisi è necessaria per il mantenimento del normale volume cellulare mediante l'espulsione di acqua e di sodio, per il mantenimento allo stato ridotto dei gruppi solfidrilici delle proteine solubili e di quelle della membrana eritrocitaria, e per mantenere il ferro in forma ridotta. L'incapacità di produrre una quantità sufficiente di energia provoca emolisi. Il glutatione, che è presente in concentrazione relativamente alta negli eritrociti, è mantenuto allo stato ridotto dall'NADPH formato attraverso la via metabolica del pentosofosfato. L'ossidazione del glutatione, provocata o da una ridotta attività glicolitica oppure direttamente dall'azione di farmaci fortemente ossidanti, provoca l'ossidazione dei gruppi solfidrilici delle proteine e quindi emolisi.
Emolisi dovuta a un difetto enzimatico eritrocitario fu rilevata per la prima volta in individui trattati con i farmaci antimalarici plasmochina e primachina. Si è poi osservato che questo difetto enzimatico è provocato dalla deficienza dell'enzima glucosio-6-fosfatodeidrogenasi (G-6-PDH). Questa è la più comune anomalia enzimatica ereditaria dell'eritrocita. Più recentemente sono stati studiati molti altri difetti enzimatici più rari (v. metabolismo dei carboidrati).
Deficienza della glucosio-6-fosfatodeidrogenasi (G-6-PDH). - L'enzima G-6-PDH catalizza la riduzione del glucosio-6-fosfato a 6-fosfogluconato e richiede il coenzima ossidato NADP+, che viene ridotto a NADPH. Solo il 10% circa del glucosio viene metabolizzato attraverso la via metabolica del pentoso-fosfato, probabilmente a causa della limitata quantità di NADP+ disponibile. Certi farmaci ossidanti aumentano la produzione di NADP+ e quindi la proporzione di glucosio metabolizzata attraverso la via metabolica del pentoso-fosfato. Se è presente un difetto enzimatico di questa via metabolica, come nel caso della deficienza di G-6-PDH, la riduzione dell'NADP+, e quindi del glutatione, è fortemente diminuita. In seguito alla somministrazione di questi farmaci ossidanti ha quindi luogo un'intensa emolisi.
La deficienza di G-6-PDH è stata scoperta durante la seconda guerra mondiale, quando il farmaco antimalarico primachina fu somministrato ai militari neri americani. Si scoprì che circa il 10% dei Negri americani sono sensibili a questo farmaco, la cui somministrazione causa una modesta emolisi, seguita da reticolocitosi (comparsa di reticolociti nel sangue circolante). La deficienza di G-6-PDH è stata osservata con frequenza molto alta in alcune popolazioni che vivono attorno al Mediterraneo - Sardi, Ebrei Sefarditi e abitanti della Turchia meridionale - e anche in Asia. La distribuzione della deficienza di G-6-PDH è simile, approssimativamente, a quella di altre anemie emolitiche ereditarie causate da difetti emoglobinici o della sintesi di emoglobina; si è per questo avanzata l'ipotesi che tale difetto enzimatico offra una certa protezione nei confronti della malaria.
Nelle popolazioni mediterranee e nei Negri americani la deficienza di G-6-PDH ha caratteristiche diverse: nella forma mediterranea si riscontra principalmente una sensibilità all'ingestione di baccelli freschi o all'inalazione di polline di piante in fiore di Vicia faba, tanto che questa anemia è chiamata anche favismo o anemia mediterranea primaverile. Anche nella condizione di sensibilità alla primachina si riscontra una deficienza dell'enzima G-6-PDH come nel favismo, ma il difetto molecolare è differente, dato che nei Negri affetti si dimostra un po' di attività enzimatica, mentre negli individui caucasici affetti l'attività enzimatica è nulla o estremamente bassa. È possibile dimostrare mediante elettroforesi in gel d'amido che le due forme dell'enzima non sono uguali e che ambedue differiscono dalla G-6-PDH degli individui normali.
L'anomalia è ereditata come un disordine genetico legato al sesso. I maschi sono quindi colpiti con una frequenza molto maggiore delle femmine, come in altre malattie ereditarie legate al sesso. Nelle femmine eterozigoti, che hanno un livello di attività enzimatica alquanto variabile, sono presenti due popolazioni eritrocitarie, una con un livello normale di enzima, l'altra con un difetto dell'attività enzimatica. La proporzione relativa delle due popolazioni eritrocitarie è variabile e dipende dal tempo e dallo stadio di sviluppo in cui l'inattivazione di uno dei cromosomi X ha avuto luogo.
La deficienza di G-6-PDH che si riscontra nelle popolazioni mediterranee è molto più grave di quella che si riscontra nei Negri americani. Crisi emolitiche gravi, tali da mettere a repentaglio la vita, seguono all'ingestione di fave fresche o hanno luogo pochi minuti dopo aver inalato polline di fava. La gravità delle crisi emolitiche era probabilmente ben nota a Pitagora e ai suoi seguaci: è noto infatti che Pitagora aveva vietato ai suoi discepoli di mangiare fave fresche e fu ucciso assieme a loro dai Crotonesi, dopo che si era rifiutato di scappare attraverso i campi di fave in fiore. Probabilmente Pitagora soffriva di deficienza di G-6-PDH; ciò fa pensare che questa forma di anemia sia abbastanza antica e che possa essere stata importata nell'Italia meridionale dai coloni greci.
Crisi emolitiche da deficienza di G-6-PDH hanno luogo non solo dopo la somministrazione di alcuni farmaci, ma anche nel corso di malattie sistemiche, come l'acidosi diabetica, oppure nelle infezioni gravi. Solo una piccola quantità di eritrociti sono emolizzati, circa il 10-15%. Poiché l'attività della G-6-PDH declina con l'invecchiamento degli eritrociti, si ammalano soprattutto le cellule vecchie; la lisi degli eritrociti è seguita da un aumento dell'attività eritropoietica e dalla produzione di cellule giovani, e ciò spiega perché la somministrazione continua di un farmaco ad azione emolitica ha progressivamente minori effetti emolizzanti. Le condizioni ossidanti che prevalgono in cellule deficienti in G-6-PDH durante una crisi emolitica provocano la precipitazione intracellulare di emoglobina: si formano corpi di inclusione e le cellule che li contengono sono rapidamente eliminate dalla milza.
Deficienza della glutationereduttasi e di glutatione. - Si tratta di due rare anomalie ereditarie, una provocata dalla deficienza dell'enzima glutationereduttasi e l'altra da un'anomalia della biosintesi del glutatione. Un'anemia moderata e ben compensata è stata osservata negli individui affetti, che sono sensibili all'azione di farmaci ossidanti.
Deficienza della piruvatochinasi. - Questa malattia è relativamente rara ed è trasmessa da un gene recessivo autosomico; gli eterozigoti sono clinicamente normali. L'enzima piruvatochinasi catalizza l'idrolisi del fosfoenolpiruvato a piruvato, con formazione di una mole di ATP per mole di piruvato formato. Gli eritrociti di individui affetti hanno un basso livello di ATP e sono incapaci di mantenere una concentrazione intracellulare di potassio sufficientemente alta; assumono inoltre forme irregolari e la loro vita media è accorciata. Una caratteristica della sindrome da deficienza di piruvatochinasi è la notevole reticolocitosi: dopo splenectomia, fino al 75% degli eritrociti circolanti nel sangue possono essere reticolociti! Queste cellule sopravvivono meglio degli eritrociti, dato che hanno ancora mitocondri e sono quindi capaci di sintetizzare efficacemente l'ATP mediante fosforilazione ossidativa.
Le manifestazioni cliniche appaiono nell'infanzia, con un grado variabile di anemia; la milza è ingrossata e si possono osservare alcune alterazioni delle ossa. La splenectomia può migliorare notevolmente la sopravvivenza degli eritrociti e diminuire la necessità di trasfusioni di sangue.
Altri difetti enzimatici causa di anemia emolitica. - Sono stati descritti nella letteratura altri rari difetti enzimatici degli eritrociti. Gli enzimi interessati sono: a) esochinasi; b) fosfoesosoisomerasi; c) triosofosfatoisomerasi; d) fosfogliceratochinasi; e) difosfogliceratomutasi. Questi difetti ereditari, osservati solo in pochi individui, sono in genere causa di anemia emolitica e, nel caso della deficienza della triosofosfatoisomerasi, di disordini neurologici ad andamento progressivo.
d) Difetti della membrana eritrocitaria.
La membrana eritrocitaria è costituita da proteine (circa il 60% in peso) e da lipidi. Le sue caratteristiche fisico-chimiche sono importanti per mantenere il corretto rapporto tra superficie e volume dell'eritrocita e la tipica forma discoidale di questa cellula. Le anomalie nella struttura della membrana provocano alterazioni della forma dell'eritrocita, e gli eritrociti deformati sono intrappolati più facilmente dalla milza e distrutti. Un'anemia emolitica cronica di gravità variabile è associata alle anomalie strutturali della membrana. Si sa ancora poco, dal punto di vista molecolare, dei difetti della membrana eritrocitaria; ciò è probabilmente una conseguenza del fatto che le nostre conoscenze sulla struttura e sulla biosintesi della membrana sono ancora relativamente scarse.
Sferocitosi ereditaria. - Questa malattia, che è la più frequente delle anomalie ereditarie della membrana eritrocitaria, viene trasmessa come un carattere dominante autosomico; essa deve il suo nome alla caratteristica forma sferica degli eritrociti (sferociti).
Gli eritrociti dei pazienti con sferocitosi ereditaria sono più suscettibili degli eritrociti normali alla lisi in soluzioni ipotoniche. La concentrazione intracellulare degli sferociti è più bassa del normale e il sodio diffonde più rapidamente in queste cellule che negli eritrociti normali. Queste osservazioni hanno suggerito l'ipotesi che responsabile della sferocitosi sia un difetto nella struttura o nella composizione della membrana eritrocitaria; la vera alterazione strutturale non è però conosciuta.
I pazienti mostrano un ittero modesto; la sferocitosi è stata anche designata come ‛ittero cronico familiare' a causa della presenza di più di un individuo affetto nella stessa famiglia. L'ittero, come altri sintomi clinici, è molto variabile, sia tra pazienti diversi, sia nello stesso paziente in periodi diversi. L'anemia e in generale di lieve entità; una complicazione frequente è costituita dai calcoli biliari, la cui formazione è provocata dall'aumentata escrezione di pigmenti biliari.
Questi sintomi sono una conseguenza dell'aumentata distruzione di eritrociti. I difetti della membrana eritrocitaria provocano l'intrappolamento delle cellule anormali nella milza: la terapia elettiva della sferocitosi è la splenectomia, dopo la quale l'emolisi cessa, anche se i difetti degli sferociti persistono e la prognosi, già relativamente buona, migliora sensibilmente.
Ellittocitosi ereditaria. - Malattia relativamente rara, ereditata come un carattere dominante autosomico e contrassegnata dalla forma ellittica della maggior parte degli eritrociti. Contrariamente a quanto si osserva nella sferocitosi ereditaria, la fragilità osmotica degli eritrociti è normale.
Non vi è correlazione diretta tra le alterazioni morfologiche degli eritrociti e la gravità dell'anemia; questa è di solito modesta e anche la splenomegalia è di lieve entità. La splenectomia è in generale di giovamento.
7. Anemie emolitiche acquisite.
Le cause delle anemie emolitiche acquisite sono in genere più eterogenee di quelle delle forme ereditarie, per cui sono possibili soltanto poche generalizzazioni. Alcune forme di anemia, provocate da deficienze nutrizionali, accorciano leggermente la vita media degli eritrociti, ma non vengono in genere classificate come forme emolitiche; per esempio, nell'anemia perniciosa vi è una diminuzione della vita media degli eritrociti in circolo, ma l'effetto senz'altro principale di tale deficienza nutrizionale è la distruzione all'interno del midollo osseo dei precursori degli eritrociti.
a) Anemie emolitiche causate da agenti infettivi.
Malaria. - La più comune causa di anemia emolitica da agenti infettivi è la malaria. Probabilmente nessun'altra malattia parassitaria ha come questa ucciso o menomato un così gran numero di individui: si è stimato che essa causi ogni anno in tutto il mondo circa due milioni di morti, metà dei quali nella sola India.
La malaria è causata da parassiti del genere Plasmodium, che sono trasmessi dalla puntura di zanzare infette del genere Anopheles. Le zanzare si infettano succhiando il sangue di individui malati: i parassiti, ancora all'interno degli eritrociti o rilasciati nel circolo sanguigno, si sviluppano nella parete dello stomaco degli insetti e migrano poi nelle loro ghiandole salivari. Le zanzare femmine prima di deporre le uova si nutrono succhiando il sangue dell'uomo, al quale trasmettono quindi i parassiti con la saliva. Si sa che quattro parassiti diversi provocano la malaria nell'uomo: P. vivax, P. ovale, P. malariae e P. falciparum. Questi parassiti non infestano i Vertebrati meno evoluti, che sono invece soggetti a una malattia simile, causata però da altri Plasmodia.
I diversi parassiti provocano forme alquanto differenti di malaria. Dopo un periodo di incubazione, di una durata compresa fra i 12 e i 40 giorni dall'inoculazione, cominciano accessi parossistici di brividi, febbre e sudorazione. Questi accessi sono provocati dall'entrata in circolo di parassiti che vengono rilasciati da eritrociti distrutti. La forma più comune di malaria è la ‛terzana', relativamente benigna, causata da P. vivax. Questo parassita si sviluppa inizialmente nelle cellule epatiche di un individuo che è stato appena infettato; solo dopo 14 giorni, quando i parassiti sono in grado di completare il loro ciclo di sviluppo intraeritrocitario di circa 48 ore, cominciano gli accessi parossistici terzanari.
All'inizio di un attacco malarico il paziente attraversa uno ‛stadio freddo' caratterizzato da brividi squassanti, intenso tremore e battito di denti. Dopo circa un'ora comincia uno ‛stadio caldo' caratterizzato da un improvviso aumento della temperatura fino a 44 °C, sensazione di malessere intenso e cefalea. Questa febbre dura circa due ore ed è seguita da uno ‛stadio di sudorazione' intensa: la temperatura cade a valori normali e il paziente si sente meglio.
P. ovale causa una forma di malaria identica alla terzana. P. malariae provoca invece la ‛quartana', caratterizzata da un periodo di incubazione che dura dai 14 ai 40 giorni e da accessi parossistici ricorrenti ogni 72 ore, che sono più lunghi e gravi di quelli della terzana.
P. falciparum è l'agente della ‛terzana maligna' o ‛malaria estivo-autunnale', una malattia più grave, che è causa della maggior parte dei decessi dovuti alla malaria. Gli accessi si manifestano irregolarmente, dopo un periodo di incubazione di 12 giorni, e sono estremamente gravi. Si distinguono diverse forme di terzana maligna: cerebrale, algida, emorragica e perniciosa. Nella forma cerebrale il delirio e il coma all'inizio della malattia sono i sintomi più caratteristici: la morte può sopravvenire in qualche ora senza che il paziente abbia ripreso conoscenza.
Nella forma emorragica, chiamata anche ‛febbre delle urine scure', le manifestazioni cliniche sono fulminanti, con rigidità, prostrazione estrema, vomito e temperatura altissima; vi è un'emolisi intravascolare massiva e passaggio di emoglobina nell'urina, per cui quest'ultima assume il colore scuro al quale la malattia deve il suo nome. La patogenesi di questa forma di malaria non è chiara. I più colpiti sono gli europei che hanno avuto ripetuti attacchi di malaria; gli attacchi si scatenano spesso in seguito all'ingestione di farmaci antimalarici, per cui si sospetta come possibile causa della malattia la sensibilizzazione agli antigeni del parassita malarico che vengono liberati in seguito all'azione dei farmaci antimalarici.
La diagnosi di malaria è basata sull'esame di strisci di sangue prelevato durante gli episodi febbrili, quando i parassiti sono visibili. La terapia preventiva della malattia si è realizzata per secoli mediante somministrazione di chinino: più recentemente ne è stata intrapresa la cura e la prevenzione con farmaci nuovi, più potenti del chinino, come la clorochina. Gli accessi parossistici vengono prevenuti dalla somministrazione di questi farmaci, ma l'eliminazione completa dei parassiti non è facilmente ottenibile e spesso la malaria appare nuovamente.
L'eradicazione della malaria dalle aree in cui è endemica si può ottenere solo eliminando le zanzare che sono portatrici dei parassiti: si devono quindi distruggere fisicamente tutti i possibili luoghi di sviluppo delle zanzare, per esempio prosciugando le paludi o cospargendole di insetticidi.
Febbre di Oroya. - È un'infezione osservata in Perù, provocata dal bacillo flagellato Bartonella bacilliformis. Viene trasmessa dal dittero Phlebotomus e provoca dolori muscolari, febbre e sensazione di freddo con rapido instaurarsi di anemia. La mortalità è alta (dal 30 al 98% dei casi).
b) Anemie emolitiche provocate da agenti chimici o fisici.
Anemie possono manifestarsi in individui esposti ad alcuni agenti chimici. Alcune sostanze chimiche provocano emolisi interferendo direttamente con il metabolismo degli eritrociti; altri agenti chimici, molto più pericolosi, possono avere un effetto emolitico iniziale, seguito poi da un'azione tossica sul midollo osseo, sfociante in un'anemia aplastica. Questa forma di anemia è caratterizzata dall'incapacità del midollo a proliferare e produrre cellule ematiche; l'anemia aplastica non è considerata un'anemia emolitica, perché la sopravvivenza degli eritrociti in circolo è pressoché normale.
Agenti chimici causa di anemia emolitica. - Diverse sostanze usate dall'industria chimica, o che venivano usate in processi di lavaggio a secco, sono causa di anemie emolitiche. Fra queste sostanze vi sono il toluene, il benzene, la fenacetina e il piombo. Le malattie provocate dall'esposizione ad agenti chimici vengono generalmente comprese nella categoria delle malattie professionali.
L'avvelenamento cronico da piombo produce un'anemia caratterizzata da microcitemia e ipocromia (diminuita concentrazione di emoglobina negli eritrociti). Il piombo inibisce l'attività di un enzima che è necessario per la biosintesi dell'eme, la sintetasi dell'acido δ-amminolevulinico. In seguito alla diminuzione della sintesi dell'eme, diminuisce anche il livello della sintesi della globina e viene prodotta una quantità insufficiente di emoglobina durante lo sviluppo degli eritrociti.
Altre sostanze chimiche e farmaceutiche possono scatenare crisi emolitiche in soggetti portatori di difetti enzimatici eritrocitari, come detto precedentemente.
Cause fisiche di anemia emolitica. - Si può riscontrare un'anemia in pazienti con ustioni estese: un'emolisi può infatti essere provocata dall'azione diretta del calore sugli eritrociti, ma anche da fattori tossici o da variazioni della composizione del plasma in seguito alla perdita di liquidi. Una forma di anemia emolitica può essere causata da danno meccanico agli eritrociti in pazienti con valvole aortiche artificiali.
c) Cause immunologiche di anemie emolitiche.
Un'anemia emolitica può essere provocata dall'entrata in circolo di anticorpi diretti contro antigeni eritrocitari. Si possono distinguere due tipi di disordini immunoemolitici: quelli provocati da anticorpi estranei all'organismo e quelli provocati da anticorpi prodotti dallo stesso paziente. Esempi tipici di anemia provocata da anticorpi estranei sono l'anemia emolitica del neonato e gli episodi emolitici acuti seguenti trasfusioni di sangue non compatibile. Le anemie provocate da autoanticorpi sono un aspetto di un gruppo complesso di disordini chiamati malattie autoimmuni, in cui il sistema immunologico del paziente perde la capacità di discriminare tra ‛l'uniforme' - i normali componenti del corpo - e ‛il non uniforme' - gli antigeni estranei (v. immunologia e immunopatologia: Malattie autoimmuni).
Anemia emolitica del neonato (eritroblastosi fetale). - Nel feto e nel neonato nei primi giorni di vita può manifestarsi una grave anemia emolitica provocata dall'immunizzazione della madre contro gli antigeni eritrocitari del feto e dal passaggio di anticorpi dalla circolazione materna a quella fetale. La causa più comune di immunizzazione è la presenza dell'antigene Rh+ negli eritrociti di un feto generato da una madre Rh-. Il gruppo sanguigno Rh è determinato da un gene autosomico complesso e il solo matrimonio che può produrre prole con anemia emolitica dovuta a incompatibilità Rh è quello di un uomo Rh+ con una donna Rh-.
La frequenza di individui Rh+ varia nelle diverse popolazioni e la frequenza dell'anemia emolitica del neonato è correlata alla frequenza di individui Rh+. Solo in una piccola parte dei matrimoni tra donne Rh- e uomini Rh+ vi è prole affetta da anemia emolitica del neonato. Il primogenito non è quasi mai colpito dall'anemia e solo da gravidanze successive possono nascere bambini affetti. L'incidenza di neonati affetti aumenta da 1:42 parti nella seconda gravidanza a 1:12 parti nella quinta gravidanza (solo i parti di donne Rh- sposate con uomini Rh+ sono inclusi in questo calcolo; la frequenza totale di anemia emolitica del neonato è di 1:200 gravidanze).
L'anemia emolitica del neonato può essere anche provocata da anticorpi diretti contro i gruppi sanguigni AB0. In questo caso non è infrequente che i primogeniti siano affetti da anemia emolitica, dato che questi anticorpi sono normalmente presenti in circolo e il loro titolo può aumentare in seguito a immunizzazione contro antigeni batterici a reazione crociata.
Questo tipo di anemia non diventa progressivamente più frequente nelle successive gravidanze, come nel caso dell'incompatibilità Rh, e sembra che tra i Negri sia la causa più comune di anemia emolitica del neonato.
La malattia può presentare gradi diversi di gravità. Nei casi più gravi il feto nasce morto, idropico e con la pelle macerata. Nelle forme meno gravi il bambino nasce ordinariamente vivo, con un ittero pronunciato che appare entro 24-48 ore e aumenta rapidamente di intensità. In casi ancora meno gravi l'ittero pronunciato può mancare del tutto, malgrado vi sia una forte anemia e il neonato appaia pallido, letargico, dispnoico e risulti cardiomegalico. In circa il 70% dei casi il bambino che soffre di anemia emolitica nasce vivo e può essere salvato da un trattamento appropriato.
Nell'anemia emolitica dovuta a incompatibilità AB0 il neonato non è in genere gravemente malato: l'ittero è di regola presente, ma l'anemia può essere minima e scompare rapidamente.
Nelle forme gravi di anemia emolitica del neonato si può osservare un quadro ematologico caratteristico. Appaiono in gran numero in circolo cellule rosse nucleate (chiamate eritroblasti), donde l'espressione ‛eritroblastosi fetale' usata per descrivere l'anemia emolitica del neonato. Queste cellule nucleate sono il prodotto di un'attiva eritropoiesi, svolta nel midollo, nella milza e nel fegato, che tenta di compensare la perdita di eritrociti distrutti dagli anticorpi. Cellule nucleate immature sono quindi rilasciate prematuramente in circolo.
La diagnosi preventiva di incompatibilità Rh è basata sulla determinazione del gruppo sanguigno di ogni donna gravida dopo il primo parto, specialmente nel caso di una donna che abbia partorito un feto morto o che abbia avuto un aborto dopo una o due gravidanze normali. Se la donna è Rh- e l'uomo Rh+ il titolo di anticorpi anti-Rh della donna deve essere misurato ripetutamente durante la gravidanza. Un aumento del titolo di anticorpi fornisce un'indicazione di immunizzazione attiva materna nei confronti del feto, che è presumibilmente Rh+. Nel caso di un'incompatibilità AB0, la determinazione del titolo di anticorpi nel siero materno ha invece pochissimo valore diagnostico.
Il grado di emolisi nel feto può essere stimato analizzando un campione di liquido amniotico; la presenza di forti quantità di pigmenti ematici è indice di un'emolisi attiva. Bisogna anche analizzare il sangue del cordone ombelicale del neonato alla nascita: un test di Coombs diretto positivo prova, senza ombra di dubbio, che gli eritrociti del neonato sono stati sensibilizzati da anticorpi. Mediante il test di Coombs è infatti possibile dimostrare la presenza di anticorpi legati a costituenti antigenici della membrana eritrocitaria: facendo reagire gli eritrociti lavati con un antisiero diretto contro immunoglobuline umane, preparato iniettando immunoglobuline umane in conigli, si osserva agglutinazione degli eritrociti ricoperti di anticorpi.
La terapia dell'anemia emolitica del neonato consiste nella exanguinotrasfusione, cioè nella sostituzione del sangue del neonato con il sangue di un donatore compatibile. Questa trasfusione deve essere portata a termine il più presto possibile quando, dopo la nascita, si riscontri un'anemia manifesta accompagnata da ingrossamento della milza e del fegato, in tutti i neonati eritroblastotici prematuri e quando il titolo di anticorpi anti-Rh della madre sia superiore a 1:64. La exanguinotrasfusione, necessaria in queste circostanze per permettere al neonato di sopravvivere, deve essere ripetuta dopo 12-24 ore se il livello della bilirubina serica sale oltre i 10 mg/100 ml. Il donatore di sangue deve essere Rh- e il suo gruppo sanguigno deve essere il più possibile compatibile sia con quello del neonato, sia con quello della madre. La quota di bambini che guariscono completamente dopo una tempestiva trasfusione supera il 90%.
È stata recentemente introdotta la profilassi dell'immunizzazione anti-Rh materna somministrando a puerpere Rh- immunoglobuline contenenti un forte titolo di anticorpi anti-Rh: questi anticorpi distruggono gli eritrociti fetali che durante il travaglio del parto sono passati nella circolazione materna. E infatti probabile che l'immunizzazione materna avvenga solo durante il travaglio e si comprende quindi come possa essere evitata dalla somministrazione di anticorpi anti-Rh, che eliminano rapidamente gli eritrociti fetali dalla circolazione materna.
Anemia emolitica autoimmune. - In questa anemia emolitica acquisita, che colpisce ambedue i sessi in qualunque età e la cui eziologia è sconosciuta, il siero dei pazienti contiene anticorpi diretti contro i loro stessi eritrociti. Mentre per la ricerca di anticorpi adsorbiti sugli eritrociti si impiega il test di Coombs diretto, già descritto, la presenza di anticorpi antieritrocitari liberi nel siero può essere dimostrata mediante il test di Coombs indiretto: gli eritrociti di un donatore con lo stesso gruppo sanguigno AB0 vengono incubati con il siero del paziente ed esposti poi a un antisiero antiimmunoglobulina; l'agglutinazione degli eritrociti così trattati rivela la presenza nel siero di anticorpi, che sono in genere del tipo ‛incompleto a caldo', mostrano attività massima a 37 °C e non fissano il complemento in vitro, pur provocando la distruzione degli eritrociti in vivo.
Sono ordinariamente rilevabili sferocitosi e splenomegalia. La splenectomia può essere di giovamento per il paziente, ma generalmente si tenta dapprima la terapia con ormoni steroidi che deprimono la sintesi di immunoglobuline e quindi degli autoanticorpi. L'impiego di questi ormoni è tuttavia limitato in considerazione degli effetti collaterali che ne conseguono, quale, ad esempio, la capacità di aumentare la suscettibilità dei pazienti alle infezioni.
Anemia immunoemolitica da farmaci. - Farmaci che si legano fortemente agli eritrociti possono provocare la formazione di anticorpi diretti contro il complesso formato dal farmaco e dalla membrana eritrocitaria e conseguentemente una forma di anemia: tra questi sono la penicillina, il chinino e la fenacetina. L'emolisi ha luogo solo dopo somministrazione del farmaco. La presenza nel siero di un paziente di anticorpi diretti specificamente contro il complesso farmaco-eritrocita può essere dimostrata incubando entrociti normali dapprima con il farmaco specifico e poi con il siero del paziente: un test di Coombs positivo ottenuto con questi eritrociti prova la presenza di anticorpi specifici.
Emoglobinuria notturna-parossistica. - Si tratta di un'anemia emolitica acquisita piuttosto rara, che si può manifestare dopo un episodio di anemia ipoplastica. L'eziologia è sconosciuta; il difetto primario è a carico della membrana eritrocitaria. Gli eritrociti divengono anormalmente suscettibili alla lisi da complemento e la loro distruzione intravascolare libera emoglobina, che passa caratteristicamente nell'urina; l'emoglobina è ordinariamente osservabile nell'urina della notte, che è più concentrata. I malati muoiono entro pochi anni, in genere di trombosi.
Anemia da agglutinine a freddo. - È un'anemia emolitica relativamente poco comune provocata da autoanticorpi che agglutinano gli eritrociti caratteristicamente a bassa temperatura. Episodi emolitici gravi possono aver luogo dopo esposizione al freddo. La malattia colpisce soggetti non più giovani, nei quali si riscontra una frequenza piuttosto alta di gammopatie monoclonali; queste sono disordini ad andamento relativamente benigno, in cui una specifica immunoglobulina, che non mostra in genere alcuna attività immunologica, è presente in concentrazione relativamente alta. In pazienti con anemia da agglutinine a freddo l'immunoglobulina monoclonale reagisce con antigeni eritrocitari.
Agglutinine a freddo sono transitoriamente presenti in pazienti con polmonite atipica primaria e con mononucleosi infettiva: esse scompaiono dopo la malattia e hanno scarso effetto emolizzante.
d) Anemie emolitiche secondarie a disordini endocrini o del sistema reticoloendoteliale.
Un'anemia emolitica può essere presente in alcuni disordini linfoproliferativi, quali la leucemia linfatica e il linfoma.
Il meccanismo attraverso il quale vengono prodotti anticorpi che reagiscono con gli eritrociti non è ancora conosciuto. Spesso il test di Coombs dà esito positivo. Il trattamento dell'anemia consiste nella terapia della malattia fondamentale.
Talvolta si può riscontrare un'anemia, generalmente di lieve entità, in pazienti che soffrono di disordini endocrini; anche in questo caso la terapia è diretta a curare il disordine fondamentale piuttosto che a trattare specificamente l'anemia.
Bibliografia.
Baglioni, C., Correlations between genetics and chemistry of human hemoglobins, in Molecular genetics (a cura di H. Taylor), vol. I, New York 1963, pp. 405-475.
Bishop, C., Surgeno, D. M. (a cura di), The red blood cell, New York 1964.
Cartwright, G. E., Diagnostic laboratory hematology, New York 19684.
Dacie, J. V., The haemolytic anaemias, voll. I-IV, New York 1954-1967.
Harris, J. W., The red cell, Cambridge, Mass., 1963.
Ingram, V. M., The hemoglobins in genetics and evolution, New York 1963.
Necheles, T. F., Allen, D. M., Finkel, H. E., Clinical disorders of hemoglobin structure and synthesis, New York 1969.
Race, R. R., Sanger, R., Blood groups in man, Oxford 1962.
Silvestroni, E., Bianco, I., La malattia microdrepanocitica, Roma 1955.
Stanbury, J. B., Wyngaarden, J. B., Fredrickson, D. (a cura di), The metabolic basis of inherited disease, New York 19662.
Weatherall, D. J., The thalassemia syndromes, Philadelphia 1965.
Wintrobe, M. M., Clinical hematology, Philadelphia 19676.
Leucemie
SOMMARIO: 1. Introduzione: a) cenni storici; b) definizione; c) classificazione; d) epidemiologia. □ 2. Eziopatogenesi: a) fattori eziologici esogeni; b) fattori eziologici endogeni; c) patogenesi. □ 3. Clinica: a) leucemia acuta; b) leucemia mieloide cronica; c) leucemia linfatica cronica. □ 4. Reperti ematologici: a) leucemia acuta; b) leucemia mieloide cronica; c) leucemia linfatica cronica. □ 5. Aspetti clinico-ematologici di forme rare: a) leucemia acuta promielocitica; b) leucemia monocitica acuta e leucemia mielomonocitica acuta; c) eritroleucemia, eritremia acuta, eritremia cronica; d) leucemia megacarioblastica; e) leucemia plasmacellulare; f) leucemia mastcellulare; g) leucemia acuta eosinofila e leucemia acuta basofila; h) leucemia acuta indifferenziata; i) leucemia congenita; l) sindrome preleucemica o dismielopoietica o mielodisplastica; m) leucemie miste; n) leucemie mieloidi croniche giovanili; o) leucemia mieloide cronica senza cromosoma Filadelfia; p) leucemia mielomonocitica cronica; q) leucemia monocitica cronica; r) leucemia neutrofila cronica; s) leucemia basofila cronica e leucemia eosinofila cronica; t) tricoleucemia o hairy cell leukemia. □ 6. Diagnosi e prognosi: a) premessa generale ed elementi di terminologia; b) leucemie acute; c) leucemie croniche. □ 7. Complicanze. □ 8. Terapia: a) concetti attuali di terapia delle leucemie acute; b) induzione della remissione completa; c) consolidamento della remissione completa; d) profilassi delle possibili localizzazioni extramidollari; e) mantenimento; f) altre terapie; g) sospensione della terapia; h) caratteristiche generali della chemioterapia; i) leucemia linfoblastica acuta; l) leucemia mieloide acuta; m) terapia di supporto; n) terapia delle recidive; o) terapia delle leucemie croniche. □ 9. Aspetti psicologici dell'assistenza al paziente leucemico. □ 10. Conclusioni. □ Bibliografia.
1. Introduzione.
a) Cenni storici.
Sebbene risalga indubbiamente a R. Virchow (v., 1845) il merito di aver esattamente individuato la natura del quadro patologico caratterizzato dall'abnorme prevalenza nel sangue di corpuscoli bianchi rispetto a quelli rossi, situazione cui egli diede il nome di weisses Blut (cioè ‛sangue bianco' o leucemia), tuttavia il primo a descrivere accuratamente un caso di leucemia fu A. Velpeau (v., 1827). A Virchow (v., 1847) risale anche il merito di aver escluso, in contrasto con J. H. Bennett (v., 1845) che contemporaneamente aveva descritto un caso simile, che l'aumento dei globuli bianchi fosse di natura piemica. Successivamente Virchow (v., 1856), oltre a chiarire il concetto di leucocitosi fisiologica e patologica, separò una forma di leucemia ‛splenica' - in cui stava in primo piano l'ingrandimento della milza - da un'altra chiamata ‛linfatica' in cui predominava l'ingrandimento dei linfonodi e in cui le cellule circolanti nel sangue, che caratterizzano la malattia, erano simili alle cellule presenti nei linfonodi. Negli anni successivi il più importante contributo fu apportato da E. Neumann (v., 1878) il quale introdusse il concetto di leucemia ‛mielogena'.
Una pietra miliare nello sviluppo delle conoscenze fu l'introduzione della colorazione panottica delle cellule sanguigne, che permise a P. Ehrlich (v., 1891) e a E. Spilling (v., 1891) di scoprire che una parte delle leucemie era caratterizzata dall'aumento del numero delle cellule granulose normalmente presenti nel sangue.
Come sempre accade nel divenire della scienza vi furono anche notevoli errori nello sviluppo delle conoscenze sulle leucemie, ma progressivamente, specie sul piano morfologico, si chiarì l'esistenza dei vari tipi di leucemia che attualmente conosciamo. Mette conto, in particolare, ricordare che A. Ferrata (v., 1912) fu l'assertore e il sostenitore della teoria monofilogenetica dello sviluppo delle cellule sanguigne, teoria che trova pieno sostegno nella ricerca più moderna la quale riconosce nella ‛cellula staminale totipotente' il precursore di tutti i leucociti (v. sangue: Organi emopoietici).
b) Definizione.
Il termine ‛leucemia', che etimologicamente significa ‛sangue bianco', indica un quadro patologico caratterizzato da un considerevole aumento del numero dei leucociti circolanti. Tale rilievo non è però assiomatico, perché l'incremento numerico dei leucociti, pur se rilevabile con grande frequenza, non lo è però costantemente, in tutti i casi di leucemia.
Si è a lungo ritenuto che le leucemie fossero ‛malattie proliferative', cioè, in sostanza, che la velocità di proliferazione delle cellule alterate che determinano in ultima analisi il quadro anatomoclinico della leucemia fosse di gran lunga superiore alla norma. È invece acquisizione abbastanza recente, ottenuta grazie alle nuove tecniche citocinetiche (v. Gavosto e altri, 1960; v. Mauer e Fisher, 1962; v. Vincent, 19743; v. Mauer, 1975), che nella leucemia la velocità di proliferazione cellulare non è superiore alla norma, anzi in effetti è spesso minore. L'anormalità fondamentale dell'elemento leucemico, specie nelle forme acute, risiede nella sua ‛incapacità alla normale maturazione'. Di conseguenza queste cellule, rimanendo immature, mantengono molto a lungo, e teoricamente in perpetuo, la capacità di proliferare, seppure a velocità ridotta. Ne consegue che le cellule leucemiche, e ciò si verifica in modo particolare nelle leucemie acute, posseggono una durata di vita e una vita riproduttiva molto lunghe: pertanto tendono ad ‛accumularsi' determinando il quadro patologico tipico dell'infiltrazione leucemica d'organo.
Si può dunque affermare che la leucemia è sostanzialmente una proliferazione atipica di uno dei tessuti che producono i vari tipi di globuli bianchi, in altri termini una neoplasia che insorge in tali tessuti o, in senso ancora più lato, una neoplasia delle cellule staminali che sono le progenitrici ditali tessuti (v. fig. 1).
c) Classificazione.
Appare ovvio dallo sviluppo della scienza che la classificazione di qualsivoglia fenomeno biologico rappresenta una richiesta della mente umana, sia per soddisfare la necessità di razionalizzazione, sia, nell'ambito della patologia umana, per possedere un mezzo utilizzabile ai fini di un'eventuale differenziazione dell'approccio terapeutico. Così nell'ambito delle leucemie umane fu dapprima riconosciuta l'esistenza di forme croniche e di forme acute, mentre l'introduzione, sul finire del secolo scorso, della colorazione panottica delle cellule permise un enorme accrescimento delle conoscenze con l'identificazione, sia nelle forme acute sia in quelle croniche, dei diversi tipi cellulari che le sostengono. Negli anni quaranta e cinquanta fu ottenuto un notevole progresso mediante l'applicazione della citochimica allo studio delle leucemie (v. Hayhoe e Quaglino, 1980).
Vengono inoltre utilizzate in questi studi le nuove acquisizioni di vari altri campi, e pertanto la classificazione delle leucemie, tranne alcune linee fondamentali per così dire storiche, è attualmente in una fase di grande progresso, specialmente per quanto riguarda le forme acute. Basterà ricordare a tale riguardo come nel simposio internazionale tenuto nel 1974 su tale argomento (v. Bessis e Brecher, 1975) siano stati messi in rilievo sia la notevole difficoltà di ottenere un accordo sulla classificazione delle leucemie acute mediante criteri esclusivamente morfologici - panottici e/o citochimici -, sia il tentativo in atto di aggirare l'ostacolo usando nuove tecniche che implicano nuovi criteri di classificazione. Quest'ultimo punto è sottolineato dal fatto che una delle tre sezioni di quel simposio era dedicata alle nuove tecniche usate nella diagnosi citologica.
Una prima semplice classificazione delle leucemie consiste nella suddivisione delle varie forme in base al loro progredire temporale (acute, croniche, subacute) e al loro rapporto con lo stipite o il tipo cellulare interessato (mieloide, linfoide, monocitico, eritroide, megacariocitico, ecc.). Una classificazione generale fondata sull'utilizzazione dei criteri suddetti, che è però suscettibile di critiche in quanto alcuni tipi considerati possono o meno essere accettati, è riportata nella tab. I.

Le metodiche utilizzate per la classificazione delle leucemie acute (LA) sono abbastanza varie. Autorevolissimi studiosi hanno sostenuto che, impiegando come unico mezzo tecnico la colorazione panottica, non sono praticamente incorsi in errore (v. Galton e Dacie, 1975). Purtuttavia è stata ed è la combinazione della colorazione panottica con le reazioni citochimiche che ha permesso di raggiungere la maggior precisione (v. Flandrin e Bernard, 1975; v. Invernizzi e altri, 1979; v. Hayhoe e Quaglino, 1980). Infatti in vaste casistiche si sono ottenuti livelli superiori al 90% di identificazione citologica delle LA (v. Flandrin e Bernard, 1975; v. Invernizzi e altri, 1979), restringendo pertanto al minimo la percentuale di leucemie acute cosiddette inclassificabili; la quota di quest'ultimo tipo è poi ulteriormente riducibile con l'impiego delle metodologie immunologiche, della determinazione dell'enzima Terminal deoxynucleotidyl Transferase (TdT; v. Bollum, 1979), in grado di identificare blasti leucemici di natura linfoide risultati di tipo inclassificabile con le metodiche citomorfologiche, e infine della tecnica delle colture in vitro.
Nel 1976 è stata proposta una classificazione delle leucemie acute su base morfologica tendente a unificare i criteri diagnostici usati da vari autori: tale classificazione, nota con l'acronimo FAB (French-American-British) perché proposta da un gruppo di ematologi dei tre paesi, ha avuto notevoli riconoscimenti ed è praticamente oggi quella di riferimento mondiale (v. Bennett e altri, 1976; v. tab. II; v. figg. 2 e 3). Essa è stata poi di recente ancor più affinata per quanto concerne i criteri da assumere per la diagnosi di certi tipi di leucemie (v. Mertelsman e altri, 1980). Occorre tuttavia ricordare che tra i vari morfologi esiste in genere una notevole concordanza nella distinzione della leucemia linfoblastica acuta (LLA) dalla leucemia mieloblastica acuta (LMA), concordanza che diminuisce notevolmente nella diagnosi dei vari sottotipi delle due forme (v. Whittaker e altri, 1979).

Occorre poi soffermarsi, seppure brevemente, sulla notevole mole di lavoro, specie su base immunologica, svolto negli ultimi lustri per migliorare le nostre conoscenze sulla natura e sull'origine delle cellule linfoidi, lavoro da cui sono scaturiti nuovi tipi di classificazione. In particolare molto importante è la suddivisione delle LLA oggi possibile in base alle conoscenze acquisite grazie all'immunologia sui vari tipi di linfociti che sono presenti nel nostro organismo (v. Brouet e altri, 1975; v. Bennet e altri, 1980; v. tab. III).

Anche nell'ambito della leucemia linfatica cronica (LLC), pur rappresentando la citochimica un notevole sussidio diagnostico (v. Bennett e altri, 1980), è ancora soprattutto il riconoscimento immunologico dei vari tipi di cellule linfoidi che ci permette una classificazione più accurata (v. tab. IV). A proposito di LLC, va poi ricordato che l'attuale suddivisione in stadi di tale malattia (v. tab. V) risulta utile nei riguardi sia della prognosi sia della terapia (v. Rai e altri, 1975).


Ancora più recenti sono le ricerche tendenti a pervenire alla tipizzazione delle cellule linfoidi su base biochimica (v. Blatt e altri, 1980); si è osservato che determinati enzimi presenti in tutti i linfociti normali, o in loro sottoclassi, sono alterati in certe neoplasie linfoidi. Un riassunto dei principali dati attualmente noti è presentato nella tab. VI.
Per quanto attiene alla leucemia mieloide acuta è verosimile che in futuro verranno proposte classificazioni fondate sulla separazione e sullo sviluppo in vitro di colonie di cellule primitive (v. Moore e altri, 1974; v. Moore, 1975; v. Spitze e altri, 1978; v. Bennett e altri, 1980).

Da ultimo va considerata l'importanza che la citogenetica ha e avrà nella classificazione delle leucemie. È ovvio che se per ogni tipo di leucemia vi fosse un marcatore nel corredo cromosomico, la classificazione e la diagnostica avrebbero risolto qualsivoglia incertezza. È interessante conoscere quale sia il punto d'arrivo delle ricerche in tale campo (v. i contributi di Rowley, 1978; v. Sandberg, 1980). È noto da tempo che nel cariogramma di circa l'85% dei soggetti affetti da leucemia mieloide cronica (LMC) è presente un tipico cromosoma chiamato cromosoma Philadelphia, simbolizzato con Ph1 (v. Rowley, 1980; v. fig. 4). Fino a qualche anno fa si riteneva il cromosoma Ph1 patognomonico della LMC: oggi è noto che esistono anche leucemie acute di tipo sia mieloide sia linfoide che presentano il Ph1 nel cariogramma (v. Rowley, The cytogenetics..., 1978 e 1980). Circa il 50% delle LA, sia del tipo mieloide sia di quello linfoblastico, presentano alterazioni cromosomiche (v. Sandberg, 1980). Purtuttavia non si è ancora in grado di elaborare quadri patognomonici e perciò classificativi basati su markers citogenetici.
Anche per le sindromi preleucemiche (v. Sokal e altri, 1980) valgono le stesse considerazioni; solo per una di esse, e precisamente l'anemia refrattaria, è stata descritta un'alterazione costante del cariogramma (v. Sandberg, 1980; v. Sokal e altri, 1980; v. genertica: Citogenetica).
d) Epidemiologia.
Le leucemie colpiscono tutto il genere umano, sebbene l'incidenza di alcuni tipi sia indubbiamente diversa nelle varie razze. Per esempio la leucemia linfatica cronica è rara nelle popolazioni orientali, mentre si manifesta con la massima frequenza tra gli Ebrei; negli Stati Uniti la popolazione bianca è più colpita di quella nera da tutte le leucemie. Differenze di mortalità per leucemia si osservano non solo tra paesi a razze diverse, ma anche tra nazioni con popolazioni razzialmente identiche (v. tab. VII); se tali differenze possono essere in parte attribuite a condizioni sociosanitarie differenti, è però verosimile che esistano reali differenze di incidenza. La tab. VII evidenzia anche una differenza di mortalità legata al sesso, che è in rapporto a una maggiore morbilità nel sesso maschile.
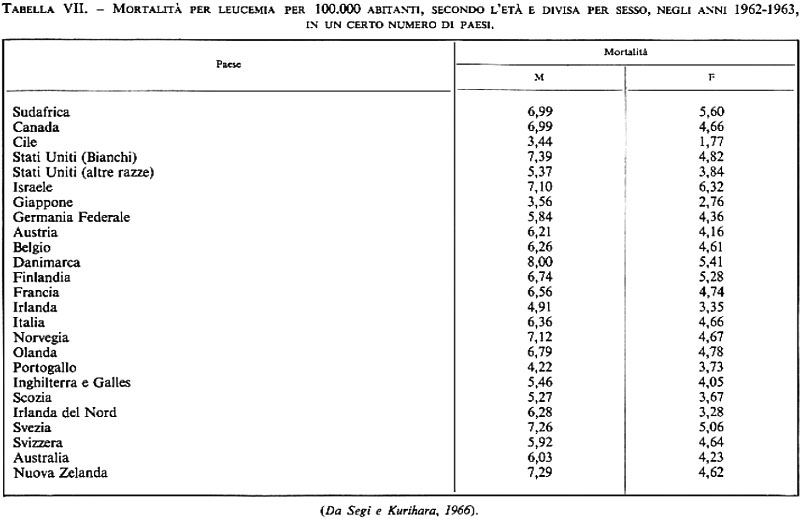
Per quanto riguarda la frequenza, le leucemie nel loro insieme occupano un posto preminente tra i tumori, pur tenendo conto della loro diversa incidenza nelle varie età: nei bambini circa la metà dei decessi per tumori è dovuta a leucemia, quota questa che scende al 20% nei giovani adulti e a valori molto più bassi (4%) negli anziani.
Per quanto attiene al tipo delle leucemie è accertata la frequenza nettamente superiore delle forme acute sulle cr0niche; queste ultime sono rare nei bambini mentre la loro frequenza, specialmente quella della leucemia linfatica cr0nica, aumenta con l'aumentare dell'età.
Rapporti chiari esistono poi tra età e incidenza delle leucemie acute: la loro frequenza mostra un primo picco entro il primo lustro di vita e di esse il 90% è di tipo linfoblastico; quindi diminuisce nettamente, per tornare a salire nella quarta decade di vita e raggiungere di nuovo alti livelli dopo i 60 anni.
Meno frequente della LLC e anche delle LA è la LMC che colpisce tutte le età, ma con frequenza diversa (v. Rizzo e altri, 1979), e si può quindi osservare, seppure raramente, anche nei bambini, con un quadro clinico-biologico identico a quello dell'adulto. Attualmente se ne conosce anche una forma detta ‛giovanile' che compare in bambini di età inferiore ai 5 anni ed è caratterizzata dall'assenza del Ph1 nel cariogramma e da alcune particolarità del quadro clinico (v. Galton, 1974). L'incidenza della LMC è massima tra i 30 e i 50 anni e mostra una netta caduta nell'età più avanzata (v. Rizzo e altri, 1979). Si conoscono rari casi di famigliarità (v. Hirschhorn, 1968).
La LLC è una malattia nettamente più frequente nell'uomo che nella donna: il rapporto di incidenza nei due sessi varia, a seconda delle casistiche, tra 1,42 e 3 in favore di quello maschile. Questo tipo di leucemia, lievemente più frequente della LMC e due volte meno frequente della LA, è tipicamente una malattia dell'età matura; risulta infatti rara tra i 20 e i 40 anni, eccezionale prima dei 20 anni e si dubita che esista nei bambini (v. Bernard e altri, 1976).
2. Eziopatogenesi.
È purtroppo d'obbligo scrivere che l'eziopatogenesi delle leucemie è ancor oggi sconosciuta, malgrado la massa di ricerche, condotte specialmente nell'ultimo trentennio, che può essere definita prodigiosa. Conosciamo alcune cause certe che inducono la leucemia sia nell'animale sia nell'uomo e si hanno probanti indizi circa possibili fattori eziologici, tra cui quello virale è sostenuto dai dati più certi, ma non si può affermare che quello che è uno dei maggiori campi di ricerca del ventesimo secolo abbia offerto la soluzione del problema eziopatogenetico di queste malattie. Schematicamente i fattori eziologici delle leucemie possono essere suddivisi in esogeni ed endogeni (v. neoplasie: Oncologia umana e Oncologia sperimentale).
a) Fattori eziologici esogeni.
Radiazioni ionizzanti. - Rappresentano il fattore esogeno leucemizzante più noto e più certo. Già nel 1930 Krebs e i suoi collaboratori dimostrarono la possibilità di indurre la leucemia in vari animali mediante irradiazione (v. Krebs e altri, 1930); ricerche più recenti hanno dimostrato la trasmissibilità da animale ad animale della leucemia indotta da radiazioni ionizzanti (v. Gross, 1959; v. Sbifrine e altri, 1971) e l'aumento dell'incidenza di leucemie nei discendenti di topine irradiate (v. Roath, 1972).
Per quanto riguarda l'uomo è stata accertata l'elevata incidenza di leucemie sia acute sia croniche tra gli individui esposti alle radiazioni ionizzanti: radiologi, minatori che estraggono metalli radioattivi, la popolazione sopravvissuta ai bombardamenti atomici di Hiroshima e Nagasaki, soggetti che per varie ragioni hanno subito terapie radianti. Proprio lo studio dei sopravvissuti di Hiroshima e di Nagasaki ha messo in evidenza che in questi individui esiste non solo un netto incremento di queste malattie rispetto alle altre popolazioni, ma anche una relazione diretta tra dose radiante ricevuta e incidenza delle leucemie, particolarmente delle forme acute (v. fig. 5; v. Watanabe, 1964; v. Tomonaga, 1962). Identiche osservazioni sono state eseguite negli Stati Uniti, in zone ove erano avvenute esplosioni nucleari (v. Lyon e altri, 1979). Anche il tipo di radiazioni ionizzanti cui si è esposti ha importanza: tra i sopravvissuti di Hiroshima e quelli di Nagasaki esposti a uguali dosi di radiazioni il rischio dell'insorgenza di leucemie è maggiore per i primi (v. Ichimaru e altri, 1978; v. Ishimaru e altri, 1979), pare per il tipo differente di radiazioni ricevute. Anche tra i radiologi l'incidenza delle leucemie, acute e croniche, era in passato molto più elevata che nel resto della popolazione: il rilievo che con una migliore protezione dalle apparecchiature radiologiche il tasso di leucemie si è drasticamente ridotto (v. Warren, 1970) costituisce una dimostrazione indiretta dell'azione leucemogena delle radiazioni. Va anche ricordata l'alta incidenza di leucemie in soggetti cui in passato era stato iniettato come mezzo di contrasto radiopaco il thorotrast, contenente l'elemento radioattivo torio: in una casistica danese sono stati riscontrati, su 756 soggetti esposti, 11 casi di leucemia manifestatisi dopo 6-30 anni dall'iniezione (v. Faber e Johansen, 1967). Il ruolo leucemogeno delle radiazioni terapeutiche non è accertato (v. Land, 1980), sebbene alcune ricerche epidemiologiche lo suggeriscano: per esempio nei soggetti irradiati per spondilite anchilosante l'incidenza di leucemie è nettamente aumentata (v. Court Brown e Doll, 1965; v. Graham, 1960; v. Polli, 1967, p. 56, per una disamina più approfondita). Pare che le radiazioni ionizzanti costituiscano un importante fattore eziologico nei confronti delle LA e della LMC, ma non della LLC (v. Bernard e altri, 1976).
Fattori chimici. Alcuni citostatici impiegati in terapia, antitumorale o meno, sembrano in grado di svolgere azione leucemogena (v. Editoriale..., 1971; v. Casciato e Scott, 1979; v. Brody e Schottenfeld, 1980; v. Rosner e Grünwald, 1980).
Il benzolo, un idrocarburo con proprietà di solvente usato in alcune lavorazioni industriali, di cui è nota l'azione aplastizzante sul midollo osseo, si comporta anche da agente leucemogeno (v. Aksoy e altri, 1972; v. Maugeri e Pollini, 1972; v. Forni e Vigliani, 1974), inducendo leucemia acuta di tipo mieloide o eritroleucemia: pertanto il suo uso è attualmente bandito.
Per altri farmaci, quali il cloramfenicolo e il fenilbutazone, mancano rilievi certi (v. Rosner e Grünwald, 1980), così come per l'acido lisergico, di cui è accertato però l'effetto lesivo sul cariotipo (v. Egozine e altri, 1968).
b) Fattori eziologici endogeni.
Fattori etnici. - La leucemia acuta è meno frequente nei soggetti americani di razza nera che in quelli di razza bianca (v. Gilliam, 1953), mentre tra i vari gruppi etnici immigrati negli Stati Uniti i Russi, i Polacchi e soprattutto gli Ebrei presentano un'incidenza di leucemie nettamente più elevata rispetto alla popolazione indigena (v. MacMahon e Koller, 1957; v. Graham e altri, 1970). L'importanza dei fattori etnici nell'eziologia della LLC è dimostrata dall'accertata rarità della sua incidenza nei Giapponesi e nei Cinesi (v. Bernard e altri, 1976, p. 1890).
Fattori prenatali. - Sono stati sospettati di essere implicati come predisponenti nell'eziologia delle leucemie vari fattori, quali la primogenitura, l'età della madre e i suoi precedenti ostetrici, e, per le femmine, il peso neonatale; nessuno di essi, però, è stato sicuramente dimostrato (ibid., p. 1853).
Fattori genetici. - Sembra accertata l'esistenza di condizioni ereditarie particolari predisponenti alla leucemia (v. Gunz, 1974). Un primo dato sicuro è che in certe famiglie possono ricorrere più casi di leucemia, molto spesso dello stesso tipo, in particolare LA e LLC (v. Bernard e altri, 1976, p. 2379). La famigliarità che talvolta si osserva nelle LA è documentata in molti lavori, tra l'altro in quello di Zuelzer e Cox (v., 1969), che prende in considerazione ben 102 famiglie. Il problema posto da tali osservazioni è stabilire se queste ricorrenze familiari delle leucemie siano dipendenti dalla casualità o se invece siano in un ben definito rapporto con l'ereditarietà. A. Videbaek (v., 1947) in un lavoro molto accurato poté concludere che i casi di leucemia familiare osservati tra 209 soggetti erano statisticamente significativi e che pertanto era ipotizzabile l'esistenza di una predisposizione all'insorgenza della leucemia. Tali risultati, tuttavia, sono stati criticati da altri autori e il problema non è ancora chiarito.
Esistono invece, come già accennato, rari gruppi familiari in cui sono presenti numerosi casi di leucemia: per essi è più facile ammettere la concorrenza genetica insieme a quella, verosimilmente, di altri fattori sconosciuti. La casistica è riportata in dettaglio da Gunz e Baikie (v. 19743, p. 70) che riportano anche quella riguardante l'insorgenza di leucemie in gemelli omozigoti per i quali sembrerebbero esistere, in base ai casi segnalati, sicuri fattori genetici. Ma anche per questi non mancano fondati dubbi. Sembra accertata, invece, l'importanza dei fattori genetici nell'eziologia della LLC: questa forma, che - come si è già detto - è assai rara in Estremo Oriente, è caratterizzata da una ricorrente incidenza familiare, tanto che non ne esiste vasta casistica ove non siano riportati casi di famigliarità (v. Bernard e altri, 1976, p., 2379). Tra l'altro va ricordato che Gunz e Fitzgerald hanno osservato in tre soggetti di una stessa famiglia affetti da LLC e in altri membri sani un'anomalia cromosomica consistente nella presenza di un cromosoma chiamato cromosoma Christchurch o CH1 (v. Gunz e Baikie, 19743, p. 152): tale anomalia è risultata tipica solo di quella famiglia e non è stata osservata in altri soggetti affetti da LLC.
Anormalità cromosomiche e leucemie. - Alcune alterazioni congenite del corredo cromosomico appaiono associate a un'alta percentuale di incidenza di leucemie acute, tanto che attualmente non si dubita che l'alterazione cromosomica giochi un ruolo importante nell'eziologia della leucemia nei soggetti che ne sono portatori. La più nota di tali associazioni si osserva nel mongolismo o sindrome di Down o trisomia G (v. Miller, 1963; v. Bass, 1971). Altre malattie genetiche comportanti un alto rischio leucemogeno sono la sindrome di Bloom, l'aplasia di Fanconi, le malattie ereditarie con fragilità cromosomica e l'atassia teleangectasica (v. Miller, 1968).
Virus. - Il problema dei rapporti tra Virus e leucemie, e più in generale tra Virus e tumori, è certamente uno dei più affascinanti e più studiati (v. neoplasie: Oncologia umana e Oncologia sperimentale). È una salda convinzione, specie dei ricercatori di base, che tali rapporti esistano realmente (v. Gallo e Meyskens jr., 1978; v. Ihle, 1978), ma purtroppo a tutt'oggi non è stata fornita la dimostrazione incontrovertibile dell'eziologia virale delle leucemie. In favore di tale ipotesi esiste però tutta una serie di dati, dall'epidemiologia alle osservazioni nell'uomo e alle sperimentazioni nell'animale. Gli studi epidemiologici hanno consentito di rilevare l'esistenza di focolai epidemici in alcune parti del mondo (v. Holland, 1970), argomento questo molto suggestivo ma estremamente controverso. I pur numerosi studi sulla trasmissione verticale delle leucemie (cioè da un genitore al figlio) non hanno fornito risultati probanti (v. Burch, 1963) né di maggiore interesse sono apparsi gli studi epidemiologici riguardanti l'eventuale insorgenza in seguito a contagio (v. Milham, 1965; v. Tjalma, 1968; v. Fraumeni jr., 1969).
È noto d'altro canto che in molte specie animali (suini, Uccelli, Rettili, Roditori, gatti, Bovini, Primati) può manifestarsi con una certa frequenza una leucemia acuta indotta - come è stato dimostrato - da Virus, tra cui quelli a RNA dotati di potere oncogeno (oncornavirus). L'eziologia virale della leucemia scoperta nel topo da L. Gross (v., 1951) è stata dimostrata anche in altri animali (v. Dutcher, 1968): in particolare è stato scoperto che il FeLV (Feline Leukaemia Virus), un virus che induce la leucemia nel gatto (v. Jarrett e altri, 1964), è presente nelle ghiandole salivari dell'animale ed è in grado di moltiplicarsi anche in tessuti umani. Accurate e approfondite ricerche non hanno però messo in luce l'esistenza di rapporti tra tale virus e la leucemia umana.
Importante è la dimostrazione, fornita in questi ultimi anni, della trasmissione tra le diverse specie animali dei virus oncogeni a RNA (v. Todaro, 1977); nell'uomo, sono stati identificati in tessuti leucemici footprints correlati a diversi gruppi di oncornavirus di tipo C (v. Gallo e Meyskens jr., 1978).
I dati più importanti in favore dell'eziologia virale delle leucemie acute nell'uomo, specie mieloidi, in quanto i risultati che riferiremo sono stati tutti ottenuti in tale tipo di LA (ibid.), si fondano sul reperto in cellule leucemiche umane (v. Gallo e altri, 1970) di un enzima chiamato ‛DNApolimerasi RNA dipendente' o ‛transcriptasi inversa', scoperto da Temin e Mizutani (v., 1970) nel virus del sarcoma di Rous e da Baltimore (v., 1970) nel virus della leucemia murina di Rauscher (v. neoplasie: Oncologia sperimentale).
Questi studi sono stati notevolmente approfonditi da Spiegelman (v., 1976) che, mediante il ‛test di rilevamento simultaneo' ideato nel suo laboratorio, un test molto sensibile per individuare la transcriptasi inversa, ha potuto dimostrare la presenza di tale enzima in cellule leucemiche umane.
c) Patogenesi.
La patogenesi delle leucemie, come la loro eziologia, rimane ancora oscura, malgrado la gran mole di recenti e rivoluzionarie acquisizioni.
Per quanto riguarda l'origine delle cellule leucemiche delle LA, in base ai risultati delle osservazioni condotte su colture in vitro di midollo di soggetti affetti da forme citomorfologicamente identificate come mieloidi (v. tab. I) è stato possibile accertare che gli elementi cellulari caratteristici di tale malattia sono di derivazione midollare, pur presentando atipie maturative (v. Moore e altri, 1974). La diversa origine delle cellule delle forme linfoblastiche è dimostrata dal diverso sviluppo delle colonie in vitro; la loro derivazione linfatica in un gran numero di casi è stata confermata da studi immunologici - basati sull'impiego di tipici markers (v. Bennett e altri, 1980) - e biochimici (v. Blatt e altri, 1980).
Non è ancora chiaro il meccanismo per cui una cellula diventa leucemica, né si conosce esattamente la natura della sua anomalia. È certo che tali cellule presentano un difetto di differenziazione ben rilevabile già con il semplice esame morfologico, difetto consistente, secondo le attuali concezioni, non tanto nella loro incapacità a differenziarsi, quanto nel carattere di parzialità del processo di differenziazione (v. Clarkson, 1972; v. Golde e Cline, 1974): ne è conferma la possibilità di riconoscere vari tipi di LA in base ai loro caratteri morfocitochimici. Il blocco maturativo delle cellule leucemiche si verifica in genere a livello di stadi alquanto precoci del processo di differenziazione, così che ne consegue un prolungamento del loro soggiorno nel midollo: tale fenomeno d'altra parte bene si accorda con l'ormai accertata ridotta velocità della loro proliferazione rispetto alle cellule normali. La ridotta differenziazione delle cellule leucemiche, ancorché associata a una ridotta proliferazione, conduce ineluttabilmente al loro accumulo nell'organismo (v. Clarkson e altri, 1970; v. Greenberg e altri, 1972).
La LA mieloide è attualmente considerata una malattia che insorge a livello della cellula staminale pluripotente (v. fig. 1); si ritiene altresì che sia una malattia clonale, caratterizzata tra l'altro dal fatto che i precursori eritroidi posseggono le stesse anomalie citogenetiche rilevate nei mieloblasti leucemici (v. Jensen e Killmann, 1967; v. Blackstock e Garson, 1974).
In altri termini il quid che induce il processo leucemico interessa sia le cellule staminali indirizzate (committed) verso la differenziazione granulocito-monocito, sia quelle indirizzate in senso eritropoietico, sia, verosimilmente, quelle indirizzate in senso megacariocitopoietico. La LA mieloide è così denominata semplicemente perché vi è accumulo e infiltrazione di cellule granulocitiche-monocitiche negli organi emopoietici, ove sono dimostrabili morfologicamente mediante biopsia. È però esperienza comune tra gli ematologi la possibilità di osservare una LA, che nella sua evoluzione sarà poi con certezza classificata come mieloide, in una fase sufficientemente precoce in cui sia giustificato formulare diagnosi di eritroleucemia o addirittura in cui le principali e più evidenti alterazioni morfologiche siano a carico della serie eritroide.
Caratteristiche delle cellule leucemiche sono anche le alterazioni cromosomiche: in quelle della LMC sono note, oltre alla presenza del cromosoma Philadelphia, detto Ph1 (v. fig. 4), numerose altre alterazioni dell'assetto cromosomico che si rendono evidenti nell'evoluzione della malattia (v. Baikie, 1966). Alterazioni del cariogramma sono rilevabili nel 50% delle LA sia mieloidi sia linfoblastiche (v. Sandberg, 1980).
Le cellule leucemiche mostrano anche varie alterazioni del patrimonio enzimatico (v. Gallo e Perry, 1968), cui attualmente non viene attribuita importanza fisiopatologica in quanto si ritiene che siano solo l'espressione di profonde alterazioni metaboliche.
Di contro, in questi ultimi anni si è registrato un notevole sviluppo delle ricerche tendenti a evidenziare ‛antigeni specifici' delle cellule leucemiche, specie nelle forme acute (v. Metzgar e Mohanakumar, 1978). Tale neoantigenicità potrebbe essere in rapporto con modificazioni della superficie cellulare indotte dall'agente eziologico (virale?): in altri termini, le leucemie acute, al pari dei tumori, insorgerebbero per l'incapacità dell'organismo di riconoscere i nuovi antigeni come estranei a se stesso e di provvedere quindi alle difese immunitarie. Si pensa che l'alterazione della ‛sorveglianza immunologica' possa giocare pertanto un ruolo importante anche nella leucemogenesi (v. immunopatologia e immunopatologia: Immunologia generale e Malattie autoimmuni).
Tra l'altro le conoscenze sull'antigenicità delle cellule leucemiche stanno alla base delle ricerche sull'immunoterapia e sul suo impiego soprattutto, ma non esclusivamente, nel trattamento delle LA.
Importanti acquisizioni di ordine fisiopatologico sono state ottenute per le LA anche mediante studi della cinetica delle cellule leucemiche (v. fig. 6), come risulta dalle ampie e recenti rassegne sull'argomento (v. Vincent, 19743; v. Mauer, 1975). Si è così potuto dimostrare che, come si è già accennato, i blasti leucemici proliferano più lentamente di quelli normali (v. Gavosto e altri, 1960; v. Mauer e Fisher, 1962; v. Gavosto e altri, 1967; v. Saunders e altri, 1967; v. Clarkson, 1969) e che buona parte di tali cellule, in una proporzione variante a seconda delle statistiche dal 40 al 90%, è in fase non proliferante, pur essendo in grado di riprendere l'attività proliferativa (v. Saunders e Mauer, 1969). Le numerose conoscenze che sono state acquisite in citocinetica hanno consentito di precisare il punto d'attacco dei vari citostatici nel ciclo cellulare e di allestire quindi precisi schemi terapeutici (v. Mauer, 1975; v. Arlin e altri, 1978; v. fig. 7; v. anche chemioterapia antineoplastica).
Nello studio dei rapporti intercorrenti tra popolazione cellulare normale ed elementi leucemici, occorre considerare separatamente la LLA e le leucemie acute non linfoblastiche (LANL). È questo un argomento della massima importanza per poter delucidare la fisiopatologia delle LA, in quanto caratteristica fondamentale ditali malattie è la sostituzione del tessuto midollare normale da parte dei blasli leucemici. Nella LLA, in base a ricerche condotte mediante colture in vitro - che dimostrerebbero come le cellule staminali in grado di dare origine alla serie granuloblastica, una volta che si induca la remissione, siano ridotte quantitativamente ma non modificate qualitativamente (v. Duttera e altri, 1973; v. Moore e altri, 1974) - è ipotizzabile che si manifesti solo una sostituzione del tessuto nobile, senza alterazione qualitativa delle tre serie midollari. Nelle LANL, il meccanismo responsabile dell'insufficienza midollare è più complesso: una serie di ricerche dimostra come le cellule staminali normali siano soppresse e/o sostituite da altre atipiche (v. Vincent, 19743).
Per quanto riguarda le leucemie croniche, occorre anzitutto notare come sembri oggi risolto il problema, per lungo tempo dibattuto, se la LMC debba essere o meno considerata una forma maligna: le moderne ricerche hanno infatti dimostrato che tale malattia è l'espressione di una iperproduzione, solo parzialmente controllata, di elementi mieloidi derivanti da cellule staminali anormali. D'altro canto la fase veramente e puramente cronica della LMC presenta tali caratteri di benignità, di ordine clinico, ematologico e di risposta alla terapia, da giustificare l'assunto di Galton (v., 1977), contestato però da Killmann (v., 1972), di considerarla una condizione preleucemica.
Come si è già detto, in oltre l'85% dei soggetti con LMC è dimostrabile il cosiddetto cromosoma Philadelphia, Ph1 (v. Rowley, 1980; v. Sandberg, 1980; v. fig. 4), che in più del 90% dei casi è conseguenza di una traslocazione tra le braccia lunghe dei cromosomi numero 22 e 9, e negli altri è il risultato di diversi e più complessi rimaneggiamenti (v. Rowley, 1980). Il rilievo che il cromosoma Ph1 è presente nel 100% delle cellule, e non solo nelle mieloidi, ma anche in quelle eritroblastiche e megacariocitiche, indica che l'anomalia risiede nelle cellule staminali pluripotenti e che un tipo alterato (eteroplastico) di tali cellule ha sostituito quelle normali. Non è ancora chiarito il ruolo del cromosoma Ph' nella fisiopatologia della LMC, se rappresenti, cioè, l'espressione del meccanismo patogenetico oppure solo un marker della LMC. D'altra parte, occorre ricordare che il cromosoma Ph1 è stato osservato anche in LA linfoblastiche e mieloblastiche, così che ne è nata una problematica attualmente tra le più dibattute (v. Rowley, 1980). Tra le alterazioni cromosomiche osservate nella LMC si deve ricordare anche la perdita del cromosoma Y, dimostrata nelle cellule leucemiche di numerosi pazienti di sesso maschile, i quali godono di una prognosi più favorevole rispetto agli altri che non presentano tale anomalia (v. Sandberg, 1980).
Numerose e importanti sono le conoscenze acquisite sulla cinetica cellulare nella LMC (v. Gavosto, 1972; v. Galton, 1974 e 1977; v. Vincent, 19743; v. Stryckmans e altri, Cell kinetics in chronic granulocytic..., 1977). Molto succintamente si può dire che in questa forma sembrano accertate la migrazione di cellule immature tra il tessuto midollare e la milza e l'importanza del ruolo svolto da quest'organo nell'iperproduzione granulocitaria, la quale rappresenta in ogni caso, anche citocineticamente, il rilievo fondamentale nella LMC. Non va però dimenticato come in questa malattia si osservino spesso anche iperproduzione di piastrine e alterazioni quantitative e qualitative, seppur più sfumate, dell'eritropoiesi. Al momento attuale è ancora completamente irrisolto il prolema rappresentato dal possibile repentino viraggio della LMC verso un quadro acuto, estrinsecantesi con varie espressioni cliniche (v. Galton, 1974 e 1977).
I grandi progressi registrati negli ultimi anni nello studio della fisiopatologia della LLC sono soprattutto dovuti alle rivoluzionarie conoscenze acquisite sui diversi caratteri immunologici dei linfociti (v. Brouet e altri, 1975; v. Bernard e altri, 1976, p. 2379; v. Brouet e Seligman, 1977) e sulla loro cinetica (v. Vincent, 19743; v. Bernard e altri, 1976, p. 2379; v. Stryckmans e altri, Cell kinetics in chronic lymphocytic..., 1977). Occorre ricordare che fino a un periodo relativamente recente un soggetto che presentava linfocitosi periferica e infiltrazione midollare linfocitica veniva catalogato come affetto da LLC. Al contrario oggi, grazie alle moderne conoscenze sulla biologia dei linfociti, si distinguono, oltre alla classica LLC, varie altre malattie, tutte estrinsecantisi con linfocitosi (v. tabb. IV e VIII). Proprio in base alla caratterizzazione immunologica dei linfociti è oggi accertato che nella stragrande maggioranza dei casi di LLC l'aumento dei linfociti circolanti è sostenuto dall'aumento dei B linfociti, rari essendo i casi in cui l'aumento dei linfociti circolanti sia dovuto all'aumento dei T linfociti (cosiddette LLC a T linfociti: v. Yodoi e altri, 1974; v. Brouet e altri, 1975; v. tab. IV).
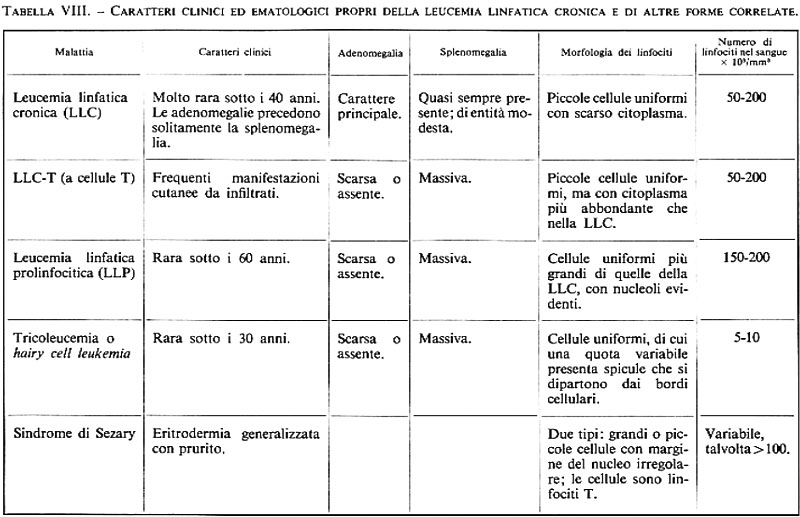
La LLC è una malattia monoclonale in cui i linfociti B sono anomali in quanto presentano difetti sia maturativi (v. Brouet e altri, 1975; v. Bernard e altri, 1976, p. 2379; v. Brouet e Seligman, 1977; v. Galton, 1977) sia della funzione secretoria delle immunoglobuline (v. Dameshek, 1967); gli studi di citocinetica hanno inoltre consentito di mettere in evidenza che in tali cellule esistono alterazioni della produzione, della durata di vita e della circolazione (v. Stryckmans e altri, Cell kinetics in chronic lymphocytic..., 1977).
Occorre infine ricordare che l'instaurarsi di una malattia eteroplastica quale la leucemia determina nell'organismo tutta una serie di alterazioni biochimiche e metaboliche molto importanti (v. Jaffe, 1975). La loro conoscenza è assai utile, sia perché esse possono essere sfruttate a scopo diagnostico, sia perché ne va tenuto conto durante il trattamento terapeutico, soprattutto per evitare l'insorgenza di complicanze che possono essere anche mortali. La tab. IX riporta le principali possibili alterazioni dell'omeostasi attualmente note.

3. Clinica.
I quadri clinici, cioè le proteiformi e talvolta caratteristiche sintomatologie con cui le leucemie si manifestano, sono l'espressione dei complessi fenomeni patologici posti alla base di tali malattie. Essenzialmente la sostituzione del tessuto midollare normale da parte delle cellule leucemiche provoca anemia, granulocitopenia e piastrinopenia, che a loro volta inducono la comparsa dei più tipici quadri leucemici. Illustreremo qui, sia per la leucemia acuta sia per le leucemie croniche, i quadri clinici classici, mentre accenneremo soltanto a quelli più rari o atipici.
a) Leucemia acuta.
L'insorgenza della malattia può essere graduale, acuta o addirittura fulminante. Il quadro clinico può inizialmente essere caratterizzato da malessere indefinito e astenia, e da quei sintomi generali che si manifestano quando si determini una condizione di anemia, qualunque ne sia l'eziologia: pallore, facile affaticabilità, acufeni, cardiopalmo, ecc. Assai di frequente sono rilevabili sintomi dovuti al deficit di piastrine, e cioè sindrome emorragica con porpora cutanea e/o mucosa, gengivorragia, epistassi, oppure presenza di veri e propri ematomi. Si è rilevato che la sindrome emorragica è presente in circa il 15% dei casi all'esordio (v. Thompson e Walker, 1962; v. Roath e altri, 1964) e in oltre il 70% durante il decorso della malattia (v. Kirshbaum e Preuss, 1943).
La sindrome emorragica può essere identificata in una vera e propria coagulopatia (v. Bernard e altri, 1976, p. 1900) così che gli ematomi possono talvolta essere molto estesi e dominare il quadro clinico, come si riscontra frequentemente nella leucemia acuta promielocitica.
La febbre è molto frequente, presente in oltre un terzo dei casi, e mostra spesso un andamento capriccioso; generalmente ne è causa un'infezione, batterica, virale o fungina (v. Levine e altri, 1974; v. Bodey, 1976): la facile recettività alle infezioni, specie batteriche, nel corso di questa malattia è in rapporto alla neutropenia. Spesso l'esordio è caratterizzato da un processo infettivo acuto che, seppure correttamente trattato, stenta a guarire. Tra le cause di febbre deve anche essere presa in considerazione l'esistenza, sia pure in una piccola percentuale di casi, della cosiddetta febbre leucemica, cioè non sostenuta da una causa infettiva ma direttamente in rapporto alla malattia primitiva, che in effetti scompare con il trattamento citostatico non comprendente steroidi.
Attualmente sono di osservazione assai rara le classiche manifestazioni a carico delle tonsille, che appaiono tumefatte, di colorito rosso cianotico, talvolta sanguinanti, ulcerate, ricoperte da membrane simil-difteriche che si estendono però oltre i limiti tonsillari. Nel cavo orale, molto spesso sede di moniliasi (mughetto) talora di rilevante entità, è anche rilevabile un'iperplasia, talvolta mostruosa, a carico delle gengive che risulta molto penosa per il paziente (v. fig. 8). Questa lesione gengivale è quasi sempre appannaggio della LA monoblastica.
Altro sintomo ben noto della LA è la dolenzia ossea, il cui riscontro è più frequente nei bambini che negli adulti (v. Thomas e altri, 1961), ma verosimilmente con una frequenza non così alta come si riteneva in passato. Comunque non è eccezionale che nell'età infantile una LA esordisca con una sindrome, protraentesi anche per vari mesi, perfettamente sovrapponibile alla malattia reumatica. Frequente è pure il dolore osseo provocabile con la pressione, specialmente allo sterno. Il valore di quest'ultimo segno è stato molto enfatizzato nel passato; esso, tuttavia, non è patognomonico della leucemia (v. McAndrew e altri, 1970), potendosi riscontrare ogniqualvolta la massa midollare è molto espansa. I segni radiologici, rari nell'adulto e assai frequenti nei bambini, oltre a un certo grado di osteoporosi frequentemente rilevabile nella LA, sono rappresentati da immagini riferibili a quattro tipi di lesioni (v. Pear, 1974): bande chiare metafisarie, non patognomoniche; lesioni osteolitiche; proliferazione del periostio; osteosclerosi. È noto che in corso di LMA possono comparire a carico specialmente del viso, ma anche di altri segmenti ossei, tumori granulocitici designati col termine di ‛cloromi' (v. fig. 9), perché anatomicamente isolati assumono una colorazione verdastra (v. Mullin e Harris, 1972; v. Pear, 1974). Questo fenomeno, tuttavia, non si verifica con costante regolarità, onde attualmente si preferisce al termine di cloroma quello di ‛sarcoma granulocitico', in quanto questi tumori sono costituiti da raccolte di cellule leucemiche granuloblastiche, o anche quello di ‛mieloblastoma' (v. Reardon e Moloney, 1961).
Anche la cute e le mucose possono essere sede di lesioni in corso di LA (v. fig. 10; v. Bleufarb, 1960). La cute è sede di alterazioni conseguenti alla diatesi emorragica - petecchie, porpora emorragica, ecchimosi, ematomi - nonché di reazioni eruttive di tipo allergico in rapporto alla somministrazione di farmaci e ai trattamenti trasfusionali (sangue in toto, piastrine, neutrofili, plasma, ecc.) cui debbono essere sottoposti i pazienti. Esistono poi lesioni cutanee dovute a infiltrazione di blasti leucemici, discretamente frequenti specialmente nella LA monoblastica e mielomonocitica e regredibili in seguito ad appropriata terapia.
L'aumento di volume dei linfonodi o adenomegalia, frequente nella LLA del bambino, lo è molto meno nella LA dell'adulto. Nelle stazioni superficiali le tumefazioni adenomegaliche, generalmente multiple, simmetriche e indolori, sono più frequentemente localizzate al collo, alle ascelle, all'inguine; le adenomegalie profonde di più frequente riscontro sono invece quelle mediastiniche, in taluni casi talmente voluminose da dar luogo a sindromi da compressione con quadri clinici drammatici, vere e proprie situazioni di grave urgenza internistica.
Specialmente nei bambini la milza aumenta di volume, in genere in modico grado, non è dolente e ha consistenza parenchimatosa elastica; più rare sono le enormi splenomegalie e gli infarti splenici, ed eccezionale è la rottura della milza (v. Ravich e altri, 1971). L'epatomegalia, che non costituisce un rilievo frequente nel quadro clinico delle LA, si osserva più comunemente nei bambini, associata alla splenomegalia.
L'interessamento renale è molto frequente secondo le statistiche anatomopatologiche, ma raramente induce sindromi cliniche e umorali (v. Sternby, 1955). Complicanze renali in corso di LA, anche così gravi da costituire un pericolo per la vita, possono invece insorgere su base iatrogena: infatti, in conseguenza della notevole distruzione cellulare provocata dal trattamento con citostatici, si determina un pericoloso aumento dei livelli uricemici, e la gran quantità di acido urico che deve essere eliminata attraverso il filtro renale può provocare la formazione di calcoli e l'instaurarsi del grave quadro della nefropatia uratica: complicanze, queste, che debbono essere assolutamente prevenute.
Le manifestazioni più frequenti a carico dell'apparato digerente sono le orali, le esofagee e le anorettali: queste ultime, in particolare, possono essere causa di gravissime sofferenze, in quanto, sia per la loro localizzazione, sia per la facilità con cui nella LA possono insorgere infezioni, facilmente si estrinsecano come ascessi a sede perianale o anorettale, con o senza fistolizzazione. Va ricordato che se nella fase di insorgenza della LA sono di rara osservazione le lesioni a carico del tubo digerente, questo nell'evoluzione della malattia è invece spesso sede di svariate complicanze, anche di natura iatrogena (v. Cornes e Jones, 1962).
Raramente partecipano al complesso sintomatologico iniziale della LA sofferenze dell'apparato cardiovascolare; sono stati tuttavia descritti alcuni casi in cui la sintomatologia iniziale era determinata dalla pericardite e talvolta dal tamponamento cardiaco (v. Battle e altri, 1969; v. Jaffe e altri, 1970). Nell'evoluzione della malattia le miocarditi e le pericarditi specifiche sono rare, mentre al riscontro autoptico si osservano infiltrati leucemici miocardici nel 37% dei casi e processi emorragici nel 54% (v. Roberts e altri, 1968). All'esordio della malattia sono di frequente riscontro alterazioni elettrocardiografiche, generalmente di significato non specifico, a eccezione di quelle di tipo ischemico conseguenti all'ipossia da anemia. Alterazioni elettrocardiografiche assai gravi sono state osservate in rarissimi casi di LA, descritti come di tipo linfoblastico con associata eosinofilia reattiva (v. Rizzo e altri, 1976): nella valutazione del significato di tali reperti occorre tener presente che sono frequentemente riscontrabili alterazioni elettrocardiografiche simili nelle cosiddette grandi eosinofilie (tipicamente nella endocardite parietale fibroplastica di Löffler). Non va poi dimenticata la cardiotossicità di molti citostatici (v. Ghione, 1977), tra cui in modo particolare la daunorubicina e l'adriamicina molto usate nella terapia delle LA (v. chemioterapia antineoplastica).
L'apparato pleuropolmonare è raramente interessato, all'esordio della malattia, da infiltrazione specifica leucemica (v. Mauri, 1978; v. i contributi di Belli e Belli, 1980), mentre molto spesso è sede di processi emorragici e infettivi di varia natura (v. Levine e altri, 1974; v. Bodey, 1976).
Per completezza si ricordano altre localizzazioni primitive della LA che sono del tutto eccezionali: testicolari, ovariche, mammarie, vescicali, ai corpi cavernosi del pene con conseguente priapismo. Occorre anche accennare alle possibili recidive della LLA nelle gonadi in soggetti giovani (v. Leef e altri, 1970), recidive di gran lunga più frequenti nei maschi che nelle femmine: attualmente si considerano le gonadi, e in special modo i testicoli, come possibili ‛santuari' (espressione mutuata dalla letteratura medica anglosassone) dei blasti leucemici, e si discute se anche esse, a somiglianza di quanto si fa ormai da anni per il nevrasse, debbano essere sottoposte a terapia profilattica (v. Harousseau e altri, 1979; v. Mauer, 1980).
Alterazioni dell'udito pare siano frequenti nel corso della LA, specie in soggetti anziani, come conseguenza di infiltrazione specifica, infezioni, emorragie (v. Shanbrom e Finch, 1958). Per quanto riguarda l'occhio, oltre alla comparsa di esoftalmo in seguito a infiltrazione ossea e/o orbitana (v. Pochedley, 1977), è possibile osservare tutta una serie di lesioni a carico dei vari segmenti (v. Rossi e Sebastiani, 1972).
Notevole importanza hanno assunto, specialmente negli ultimi anni, in rapporto alla più lunga sopravvivenza dei pazienti, le manifestazioni neuromeningee in corso di LA. Se infatti esse sono rare in fase di esordio, sotto forma per solito di emorragia cerebrale - in genere repentinamente fatale - o di meningite, divengono più frequenti nel decorso della malattia per l'alta percentuale di pazienti, specialmente di quelli affetti da LLA in età infantile, la cui sopravvivenza si protrae per anni in seguito a una terapia ben condotta (v. Hyman e altri, 1965; v. Fontana e altri, 1975 e 1978; v. Fontana e Rizzo, 1976; v. Montuori, 1977; v. Pochedley, 1977). Le complicanze neuromeningee sono molteplici e proteiformi: emorragie, trombosi, processi infettivi, infiltrazioni leucemiche a livello meningeo, cerebrale e spinale, alterazioni iatrogene (conseguenti all'impiego di citostatici, all'esecuzione di rachicentesi o al trattamento con radiazioni ionizzanti). Le emorragie, che possono rappresentare la causa dell'exitus, sono attualmente di osservazione non molto frequente; la loro causa può anche essere iatrogena (v. Priest e altri, 1980). I rari processi infettivi conseguono generalmente a stati setticemici insorgenti per l'ipoplasia midollare determinata dalla fase di induzione della terapia citostatica (v. cap. 8) o, ancor più raramente, a penetrazione di agenti infettivi in corso di rachicentesi (v. Fontana e altri, 1975; v. Bernard e altri, 1976, p. 1890).
L'infiltrazione leucemica del nevrasse, che si manifesta per lo più con il quadro della meningite leucemica, rappresenta un problema di grande attualità. Mentre in passato, quando il decorso della LA era estremamente rapido, tali complicanze si osservavano in circa il 20% dei pazienti, attualmente, con la sopravvivenza notevolmente prolungata per effetto delle efficaci terapie disponibili, esse si manifestano nel 50-60% dei soggetti affetti da LA e non sottoposti alla profilassi delle complicanze stesse (v. Evans e altri, 1970). Tale fenomeno si può facilmente spiegare tenendo conto che il nevrasse è avvolto dalle meningi, che sono ‛impermeabili' ai citostatici correntemente impiegati per la cura delle LA; esso quindi rappresenta un ‛santuario' per le cellule che, sia pure in numero limitato, lo abbiano già infiltrato al momento dell'inizio della terapia. Da tali osservazioni è derivata l'attuale ‛terapia profilattica' del nevrasse, basata sull'irradiazione dell'encefalo e sull'introduzione di citostatici nello speco vertebrale mediante rachicentesi: questo schema terapeutico è stato standardizzato dagli ematologi del St. Jude Children's Research Hospital di Memphis, Tennessee (v. Simone, 1974; v. Mauer, 1980).
Anche nelle LMA alla prolungata sopravvivenza ha corrisposto l'aumento di frequenza delle complicanze meningee, tuttavia inferiore a quello che si osserva nel decorso della LLA, per ragioni senza dubbio molto complesse. La meningite leucemica può manifestarsi precocemente nel decorso della malattia, oppure a distanza di anni, talvolta di molti anni, dalla remissione (v. Bernard e altri, 1976, p. 1890). L'insorgenza di tale complicanza precede molto spesso la ricaduta ematologica: tra i due episodi l'intervallo di tempo varia, ma mediamente è di alcuni mesi. In casi molto rari è stato osservato che a un episodio meningitico, insorto poco tempo dopo la remissione ematologica, ha fatto poi seguito una remissione neurologica molto lunga (v. Schaison e altri, 1973). La diagnosi della complicanza si formula generalmente in base al reperto dei blasti leucemici nel liquido cefalorachidiano (v. Fontana e altri, 1978; v. fig. 11), in quanto i soggetti malati vengono regolarmente sottoposti a rachicentesi ai fini del trattamento profilattico e del controllo. Il reperto, talvolta limitato a poche cellule attualmente evidenziabili grazie all'impiego di nuovi mezzi tecnici (v. Fontana e altri, 1978), è molto spesso disgiunto da qualsivoglia sintomatologia soggettiva e obiettiva; in altri casi, invece, è accompagnato dal classico corteo sintomatologico dell'ipertensione endocranica, rilevabile con i consueti metodi semeiologici fisici e strumentali. Appare pertanto indispensabile non sottovalutare i sintomi, anche apparentemente banali, che il paziente può presentare durante una documentata remissione clinico-ematologica. La diagnosi precoce dell'interessamento del sistema nervoso centrale sarebbe formulabile attualmente anche con metodi biochimici (v. Mavligit e altri, 1980).
Le infiltrazioni dell'encefalo possono provocare convulsioni, emiplegia, turbe psichiche, diabete insipido, nonché una sindrome di obesità con polifagia regredibile in seguito a opportuna terapia (v. Fontana e altri, 1973; v. fig. 12).
Altre manifestazioni neurologiche osservabili in corso di LLA sono le lesioni a carico dei nervi cranici, nella maggior parte dei casi unilaterali, e le paralisi multiple: queste alterazioni, suscettibili di regressione completa in seguito a una terapia ben condotta, costituiscono talvolta il primo segno dell'emopatia. Infine occorre tener presente che il trattamento stesso della LA può essere causa di complicanze neurologiche (v. Weiss e altri, 1974), che pongono spesso difficili problemi di diagnostica differenziale.
b) Leucemia mieloide cronica.
La LMC è caratterizzata da una prima fase di stasi, cronica, cui fatalmente, dopo un intervallo di tempo differente da caso a caso, segue una fase variamente definita come acuta, di metamorfosi, accelerata.
La possibilità di diagnosticare una LMC del tutto casualmente in fase completamente asintomatica è, da alcuni anni, divenuta più frequente in conseguenza dell'automatizzazione dell'esame emocromocitometrico e della sua conseguente estensione, come normale routine, a tutti i soggetti che per qualunque motivo si sottopongono a un prelievo di sangue (v. Galton, 1974). I sintomi generali più comuni che inducono il paziente a consultare il medico sono l'astenia, il pallore, la dispnea, la facile esauribilità, il calo ponderale, l'inappetenza, la precoce sensazione di pienezza di stomaco seguente al pasto, la difficoltà della digestione. Altre volte i sintomi iniziali sono in rapporto alla splenomegalia: senso di peso o di ingombro all'ipocondrio sinistro, rilievo accidentale dell'esistenza di una massa addominale. Assai più raramente il quadro clinico è aperto da una sindrome emorragica, da trombosi, da dolori all'epigastrio cui molto spesso corrisponde il riscontro di un'ulcera gastrica (v. Wintrobe, 19747, p. 1500), da dermopatie sostenute da infiltrati leucemici, da artralgie, da artropatia gottosa acuta, da dolori ossei, da priapismo negli uomini (raramente osservato anche in bambini) e da crisi clitoridee nelle donne, da febbre certamente non settica e suscettibile di remissione dopo terapia specifica per la LMC, da infarto splenico, da una sindrome similtireotossica. In casi ancora più rari la sintomatologia iniziale è molto simile a quella della LA e comprende dolori ossei, sindromi infettive o emorragiche, adenomegalie mono- o pluristazionali per metaplasia mieloide dei linfonodi. Tra queste atipiche manifestazioni iniziali vanno, inoltre, annoverati i sarcomi mieloidi, che possono insorgere in varie parti del corpo, e le fratture ossee patologiche.
Come abbiamo già detto, quando la malattia è scoperta in uno stadio molto precoce può mancare ogni reperto obiettivo. Una volta che essa si sia resa evidente, l'obiettività classica della LMC è rappresentata dalla splenomegalia, dal pallore e dalla sternalgia; rilievi più rari sono una modica epatomegalia e manifestazioni emorragiche di entità assai varia (dalla porpora appena pronunciata ai grossi ematomi, alle emorragie retiniche). L'entità della splenomegalia è varia: dal modico aumento di volume, che rende il polo inferiore dell'organo appena palpabile, all'enorme tumore di milza (v. fig. 13) i cui diametri verticale e trasversale si sviluppano rispettivamente nello scavo pelvico e oltre la linea addominale xifo-ombelicale: quest'ultimo tipo di splenomegalia costituisce un frequente e tipico riscontro di altri processi patologici, quale la mielofibrosi idiopatica. Generalmente la splenomegalia della LMC è di discreta entità, tale che il polo inferiore dell'organo è palpabile molti centimetri (10 in media) al di sotto del margine costale (v. Wintrobe, 19747, p. 1500).
Come si è già detto, nella LMC alla fase cronica fa seguito, in un'altissima percentuale di casi (70-80%), una chiara e netta modificazione bio-ematologica della malattia. Questa nuova fase, che può essere definita genericamente di ‛metamorfosi', si estrinseca in vari modi: più frequentemente con precipue stigmate ematologiche, identificabili in ‛crisi blastica' e in ‛trasformazione acuta', assai più raramente con stigmate cliniche e/o ematologiche più sfumate o generiche.
L'esordio di questa seconda fase è frequentemente caratterizzato da sintomi generali quali astenia talora profonda, febbre, dimagramento, sudorazione notturna; più raramente il paziente accusa artralgie, dolori ossei, cefalea in rapporto a interessamento meningeo. Tra le manifestazioni più rare vanno ricordate le lesioni osteolitiche, che possono anche causare fratture patologiche (v. Rizzo e altri, 1977).
I più frequenti rilievi obiettivi sono rappresentati dalla splenomegalia ingravescente, o comunque non più controllabile con la consueta terapia, dal pallore, dallo scadimento delle condizioni generali, dalla ricomparsa della sternalgia e/o dalla comparsa di dolorabilità ossea diffusa alla pressione, dalle adenomegalie, dall'epatomegalia.
c) Leucemia linfatica cronica.
Anche la LLC in numerosi casi (25% secondo Galton: v., 1966) viene diagnosticata incidentalmente. I disturbi più frequentemente accusati dai pazienti sono quelli dipendenti dallo stato di anemizzazione (pallore, dispnea, vertigini, acufeni, fenomeni anginosi, abbastanza frequenti, questi ultimi, in quanto la malattia è prevalente nei soggetti anziani), la facile affaticabilità, o la comparsa di adenomegalie. Più raramente si manifestano diatesi emorragica secondaria alla piastrinopenia, oppure episodi infettivi che stentano a guarire, disturbi in rapporto alla splenomegalia, che talvolta può accidentalmente essere percepita dallo stesso paziente o dolori ossei.
I reperti obiettivi più frequenti sono rappresentati dalle adenomegalie, dalla splenomegalia e dalla sternalgia provocata con la pressione, che si riscontra però con una frequenza nettamente inferiore rispetto alla LMC: tutti questi sintomi sono in rapporto all'infiltrazione delle cellule leucemiche. I linfonodi sono modicamente aumentati di volume, non dolenti né adesi ai tessuti vicini. La splenomegalia è modesta: in genere la milza deborda meno di 10 cm dall'arcata costale. Nella LLC, l'infiltrazione cellulare leucemica è ubiquitaria e può manifestarsi in vari distretti: nella pelle, sotto forma di chiazze o noduli di colore rosa, marrone o purpureo; nel fegato, che aumenta di volume e causa quindi senso di peso o dolori all'ipocondrio destro; nel tubo digerente, specialmente a livello dello stomaco e del piccolo intestino, determinando una sintomatologia molto varia, dalla sindrome dolorosa alla diarrea, all'emorragia discreta o grave, al malassorbimento. L'ingrandimento massivo dei linfonodi, specie in certe sedi, può dar luogo a situazioni cliniche particolari, che richiedono interventi terapeutici urgenti: così quello dei linfonodi mesenterici può determinare occlusione intestinale, quello dei linfonodi retroperitoneali può causare la compressione bilaterale degli ureteri con conseguente anuria. L'apparato respiratorio può essere interessato da infiltrati alle tonsille, la cui tumefazione in rari casi è responsabile di una sindrome ostruttiva alta, ai polmoni, ove si presentano diffusi con aspetto nodulare o miliare, oppure alle pleure con conseguente versamento.
Il sistema nervoso centrale pare scarsamente interessato nella LLC (v. Hansen, 1978). Nello scheletro durante il decorso della malattia è in genere dimostrabile l'osteoporosi, mentre rare sono le lesioni di tipo osteolitico (v. Pear, 1974; v. Rizzo e altri, 1977). I reni, in base a statistiche necroscopiche, risultano interessati in oltre il 60% dei casi di LLC: fortunamente questi dati non coincidono con quelli clinici, in quanto in pratica il riscontro di manifestazioni urologiche importanti in corso di LLC è piuttosto raro.
La LLC è una malattia a lento decorso, protraentesi per anni, la cui base fisiopatologica è costituita dall'accumularsi nell'organismo di piccoli linfociti che proliferano lentamente. Tale lento sviluppo della malattia è responsabile di una progressiva modificazione della sintomatologia soggettiva e obiettiva. Così la progressiva infiltrazione del midollo osseo da parte dei linfociti, con riduzione delle normali cellule midollari, determina la diminuzione degli eritrociti, dei granulociti e delle piastrine nel sangue periferico: conseguentemente nel decorso della malattia si manifestano anemia, con i relativi segni e sintomi, diatesi emorragica e tendenza alle infezioni, determinata dal deficit dei neutrofili e in parte anche dalle profonde modificazioni, indotte dalla LLC, delle funzioni immunologiche dell'organismo, tra cui l'alterazione della produzione di immunoglobuline.
Per quanto concerne altre forme rare di malattie linfoproliferative croniche, che nella trattatistica sono accomunate alla LLC, ne abbiamo riassunte le principali caratteristiche cliniche nella tab. VIII.
4. Reperti ematologici.
Lo studio del sangue periferico e del midollo osseo è di importanza essenziale per la diagnostica delle leucemie acute e croniche. Il sangue periferico si preleva generalmente mediante puntura di un polpastrello, il midollo osseo si ottiene invece con la tecnica della mielobiopsia praticata in punti precisi dello scheletro: la spina tibiale nei bambini molto piccoli, la spina iliaca posterior-superiore nei bambini più grandicelli, la parte centrale dello sterno o la spina iliaca posterior-superiore o la cresta iliaca o molto raramente altre sedi nell'adulto. Sia il sangue periferico, sia il midollo osseo vengono strisciati su vetrini: i preparati così ottenuti sono poi sottoposti a colorazione (di May Grünwald-Giemsa, o altra equipollente), in modo che, in base alla loro differente affinità tintoriale, le cellule possano venire chiaramente studiate e classificate al microscopio.
a) Leucemia acuta.
Nella LA è reperto caratteristico la presenza nel sangue periferico di cellule atipiche chiamate ‛blasti leucemici'. In più del 50% dei casi vi è leucocitosi, che tuttavia non rappresenta una conditio sine qua non per la diagnosi di LA: in una discreta percentuale di casi il numero dei leucociti è compreso nei limiti normali o è addirittura inferiore a questi (v. tab. X). Ferrata coniò il termine, mantenuto ancora attualmente da Wintrobe (v., 19747, p. 1482), di ‛leucemia aleucemica' per indicare quei casi di LA in cui nel sangue periferico non sono reperibili con le comuni metodiche blasti leucemici. In realtà tali cellule sono presenti in circolo, ma in numero così esiguo che possono essere dimostrate e studiate, anche citochimicamente, solo impiegando la tecnica, peraltro semplice e di facile esecuzione, dell'arricchimento del sangue periferico.

Reperto tipico nel sangue periferico e nel midollo osseo di soggetti affetti da LA è lo hiatus leucemicus, consistente nella presenza di cellule altamente immature accanto ad altre mature, senza forme intermedie di passaggio.
Nel midollo osseo è dimostrabile l'infiltrazione di cellule leucemiche con scomparsa o forte diminuzione delle normali cellule midollari (v. figg. 2 e 3). Il numero complessivo delle cellule presenti nel midollo è in genere aumentato, ma in alcuni casi può essere normale o addirittura inferiore alla norma, condizione, questa, definita di ‛midollo ipocellulare'. Talvolta i tentativi di ottenere materiale midollare con l'usuale tecnica della mielobiopsia effettuata in diversi punti dello scheletro risultano infruttuosi, dando luogo alla cosiddetta punctio sicca: in tali casi si procede, mediante un particolare ago tipo Jamshidi, alla osteomielobiopsia. Il frammento di tessuto cosi ottenuto permette lo studio istologico del midollo osseo, ed è allora talvolta possibile dimostrare come il midollo abbia ricca cellularità associata a una fibrosi che è causa della punctio sicca. Tali situazioni pongono problemi molto impegnativi di diagnostica differenziale, per esempio tra LA e aplasia midollare, processi morbosi per i quali la prognosi e la condotta terapeutica variano notevolmente.
Un reperto che ancora oggi conserva il suo carattere patognomonico, nella diagnosi di LA non linfoblastiche, è quello della presenza nei blasti dei bastoncini di Auer (v. fig. 14), inclusi generalmente di forma allungata bastoncellare, talvolta rotondeggiante, colorabili in rosso nelle comuni preparazioni; singoli o multipli, in alcuni casi riuniti a ciuffo, i bastoncini di Auer si originano come conseguenza dell'alterazione della maturazione della cellula leucemica mieloide, nella quale i granuli primitivi, chiamati ‛granulazioni azurofile', anziché rimanere isolati come nei normali mieloblasti e promielociti, confluiscono (v. Laszlo e Rundles, 19772). Nelle LA sono poi comuni altri reperti ematologici, non specifici, secondari alla diminuzione delle cellule midollari progenitrici, come la riduzione del tasso di emoglobina, di eritrociti e di piastrine, e alterati valori ematochimici (v. tab. IX), alcuni comuni a tutti i tipi di LA (per es. l'iperuricemia), altri specifici di alcune forme (per es. l'aumento del lisozima nel sangue e nelle urine in corso di LA monoblastica).
b) Leucemia mieloide cronica.
Nella fase cronica è caratteristico il reperto di un elevato numero di leucociti nel sangue periferico, fino a oltre 100.000/mm3: sono noti casi in cui al momento della diagnosi la leucocitosi raggiungeva valori di 500.000 cellule per mm3 (v. Rizzo e altri, 1979). La formula leucocitaria consente di dimostrare la presenza in circolo, oltre che di un numero fortemente aumentato di neutrofili, anche di cellule mieloidi immature (v. Rizzo e altri, 1977; v. fig. 15) con prevalenza di mielociti e metamielociti, nonché l'aumento di basofili, eosinofili e monociti. Il midollo osseo appare caratteristicamente ipercellulare, con iperplasia netta della serie mieloide, molto ricca di mielociti e metamielociti; in esso è anche dimostrabile un aumento degli elementi delle serie eosinofila e basofila e dei megacariociti, molti dei quali appaiono di volume nettamente ridotto. Nel midollo di soggetti affetti da LMC è anche possibile osservare cellule simil-Gaucher e istiociti blu mare (v. Lee ed Ellis, 1971), identificabili come elementi tesaurismotici.
Al momento della diagnosi in alcuni pazienti può essere messa in evidenza anemia, piastrinopenia o piastrinosi. La drastica riduzione della fosfatasi alcalina leucocitaria, di cui i neutrofili sono ricchi, nella maggior parte dei casi di LMC (v. Wachstein, 1946) costituisce un reperto di notevole importanza per la diagnosi. Tra i reperti ematochimici è molto frequente la condizione di iperuricemia e sono possibili quelle di iperkaliemia, ipoglicemia, aumento dell'istamina, aumento del livello serico di vitamina B12.
Nella fase acuta della LMC nel sangue periferico e nel midollo si osserva, a grandi linee e con varia modalità di insorgenza e di evoluzione temporale, il quadro ematologico della LA (v. fig. 16). Anche in questa fase, come nella LA, la progressiva scomparsa dal midollo delle cellule progenitrici normali causa la diminuzione dell'emoglobina, degli eritrociti e delle piastrine, mentre il reperto di un netto incremento di basofili nel sangue circolante è molto frequente ed è possibile la presenza di micromegacarioblasti. La fosfatasi alcalina leucocitaria, sensibilmente ridotta o assente nella fase cronica della malattia, aumenta nella fase acuta.
Recenti contributi sulla crisi blastica della LMC hanno fornito l'importantissima dimostrazione che in circa il 10% dei casi le cellule blastiche leucemiche sono di tipo linfoide (v. Spiers, 1979), vale a dire che una leucemia cronica di tipo mieloide può esitare in una lucemia acuta di tipo linfoide. L'accertata validità di tale acquisizione conferma la teoria secondo la quale nella LMC la noxa patogena agirebbe a livello della cellula staminale totipotente, in grado di dare origine sia alle cellule mieloidi sia a quelle linfoidi, inducendone la trasformazione neoplastica.
c) Leucemia linfatica cronica.
In questa forma il reperto tipico nel sangue circolante è un netto aumento del numero dei leucociti, con valori però solo raramente superiori a 100.000/mm3, il 70-90% dei quali è costituito da linfociti: v'è quindi una differenza con quanto si osserva nella LMC, in cui mediamente la leucocitosi al momento della diagnosi risulta più elevata. Come è noto, la frazione percentuale di linfociti è direttamente proporzionale all'entità della leucocitosi e comunque, se a leucocitosi non molto elevata corrisponde linfocitosi assoluta, è raro che a leucocitosi anche spiccatissima corrisponda neutropenia assoluta. È stato già segnalato che attualmente la LLC è suddivisa in stadi (v. tab. V) cui corrispondono diversi indici di sopravvivenza (v. Phillips e altri, 1977).
Al microscopio ottico i linfociti della LLC mostrano caratteristiche morfologiche simili a quelle dei piccoli linfociti normali (v. fig. 17); l'esame di strisci di sangue periferico consente con grande frequenza di dimostrare la presenza, tipica di questa forma, di frammenti nucleari, le cosiddette ombre di Gumprecht, espressione della particolare fragilità dei linfociti. Sul piano morfologico è possibile operare una distinzione, in base ai caratteri cellulari, tra LLC e altre forme linfoidi croniche (v. tab. VIII). Dal punto di vista citochimico questi linfociti sono caratterizzati dalla presenza di granuli più o meno voluminosi positivi alla reazione al paS, specifica dei mucopolisaccaridi.
Al livello del midollo osseo, l'infiltrazione di piccoli linfociti senza caratteri di atipie costituisce il reperto peculiare della LLC (v. fig. 18). Convenzionalmente l'infiltrazione si considera significativa quando la quota dei linfociti rappresenta almeno il 40% della popolazione cellulare midollare; l'esperienza insegna che vi è netta correlazione tra linfocitosi periferica e infiltrazione linfocitaria midollare. Al momento della diagnosi un discreto numero di pazienti presenta un modico grado di anemia e di piastrinopenia.
Molto frequenti nella LLC sono i disordini immunitari, che influenzano in modo grave sia la sintomatologia sia l'evoluzione della malattia (v. Cone e Uhr, 1964): in un notevole numero di casi una condizione di ipogammaglobulinemia, che rende conto della facilità con cui i soggetti affetti da LLC contraggono le infezioni batteriche, è dimostrabile già al momento della diagnosi o lo diviene durante l'evoluzione della malattia. L'alterazione dell'immunità cellulare, o ritardata (v. immunologia e immunopatologia: Immunologia generale), che si mantiene normale nei confronti di antigeni conosciuti e deficitaria contro nuove stimolazioni antigeniche, spiega la facilità dell'insorgenza delle infezioni virali in tali pazienti. In discrete percentuali di pazienti durante il decorso della LLC è dimostrabile la produzione di immunoglobuline monoclonali o di crioglobuline, oppure la comparsa di fenomeni autoimmunitari responsabili di ben note complicanze, quale l'anemia emolitica (v. immunologia e immunopatologia: Malattie autoimmuni).
5. Aspetti clinico-ematologici di forme rare.
a) Leucemia acuta promielocitica.
Descritta per la prima volta da L. K. Hillestad (v., 1957), è stata particolarmente ben studiata dal punto di vista morfologico e clinico e nelle sue possibilità terapeutiche da J. Bernard.
Il carattere ematologico di questa forma è costituito dalla particolare ricchezza di granulazioni dei blasti leucemici (v. fig. 3), mentre tra i rilievi cimici domina la diatesi emorragica che si manifesta tipicamente sotto forma di grandi ecchimosi in rapporto alla coagulopatia intravascolare disseminata (CID) che con grande frequenza complica questo tipo di LA.
b) Leucemia monocitica acuta e leucemia mielomonocitica acuta.
L'esatta posizione nosografica di questi tipi di LA è stata per lungo tempo oggetto di discussione: recentemente i risultati di un approccio multidisciplinare sembrano aver consentito con sicurezza la loro sistemazione nel gruppo delle LA di tipo mieloide (v. Straus e altri, 1980).
Clinicamente è caratteristica la frequenza dell'infiltrazione leucemica tissutale responsabile, nei vari casi, di epatosplenomegalia, di ipertrofia gengivale (v. fig. 8), di manifestazioni cutanee, di adenomegalia, di meningite leucemica (ibid.). Il reperto ematologico caratteristico è rappresentato dalla monocitosi, dimostrabile con le colorazioni panottiche e/o con le reazioni citochimiche.
c) Eritroleucemia, eritremia acuta, eritremia cronica.
Queste malattie, individuate e descritte da G. Di Guglielmo (v., 1923 e 1962), sono riunite in un gruppo designato, secondo la terminologia proposta per la prima volta dall'americano W. Dameshek, ‛sindrome di Di Guglielmo'. I reperti peculiari dell'eritroleucemia e dell'eritremia acuta sono individuabili solo sul piano ematologico, in quanto il loro quadro clinico coincide con quello delle LA.
Nell'eritroleucemia la noxa patogena induce la trasformazione neoplastica sia delle cellule mieloidi sia di quelle eritroidi, così che lo studio morfologico del midollo osseo consente di dimostrare la presenza di blasti leucemici mieloidi e, a carico degli elementi eritroblastici, di alterazioni di tipo quantitativo, morfologico (atipie, aspetti megaloblastoidi, poliploidismo) e qualitativo (reazione positiva al paS, anziché negativa come si osserva negli elementi entroblastici normali).
In una percentuale nettamente inferiore di pazienti le alterazioni leucemiche blastiche interessano solo e permanentemente le cellule della serie eritroide: tali casi il più delle volte presentano un andamento acuto (eritremia acuta) e solo rarissimamente quello cronico, con sopravvivenza di anni (eritremia cronica).
d) Leucemia megacarioblastica.
L'esistenza di questo tipo di LA, in passato molto discussa, è attualmente accertata grazie soprattutto ai risultati delle ricerche di citochimica ultrastrutturale. La malattia è essenzialmente caratterizzata dalla fibrosi midollare, dalla somiglianza rilevabile con la colorazione panottica tra questi blasti leucemici e i linfoblasti e dal decorso clinico rapidamente fatale per scarsa sensibilità alla chemioterapia (v. Bréton-Gorius, 1979).
e) Leucemia plasmacellulare.
È questa una rara forma di esordio del mieloma multiplo, le cui caratteristiche patologiche e cliniche consentono, almeno secondo alcuni autori, di considerarla un'entità morbosa a sé stante, inquadrabile tra le LA (v. Woodruff e altri, 1978). Dal punto di vista clinico i rilievi più importanti e frequenti sono l'epatomegalia e l'adenomegalia associate, come nel mieloma multiplo, alle lesioni osteolitiche e all'infiltrazione precoce di vari tessuti. Ematologicamente il reperto patognomonico è rappresentato dall'infiltrazione del midollo osseo da parte di plasmacellule atipiche che, al contrario di quanto avviene nel mieloma multiplo a espressività classica, passano anche in circolo e sono osservabili nel sangue periferico. La prognosi è in genere molto severa per la scarsa sensibilità alla chemioterapia.
f) Leucemia mastcellulare.
Forma estremamente rara (J. Bernard nella sua casistica di oltre 1.400 casi di LA ne riporta un solo caso), senza un quadro clinico uniforme e ben definito, ematologicamente caratterizzata dalla presenza nel sangue periferico e dalla grave infiltrazione midollare di mastociti leucemici (v. Coser e altri, 1980).
g) Leucemia acuta eosinofila e leucemia acuta basofila.
Si fa qui appena menzione di questi due tipi di LA, la cui esistenza è fortemente messa in dubbio dalla più recente trattatistica mancandone sicuri presupposti biologici.
h) Leucemia acuta indifferenziata.
Come abbiamo già detto (v. cap. 1, È c) il numero dei casi di questa LA si è progressivamente ridotto con l'affinarsi e il moltiplicarsi dei mezzi tecnici che hanno consentito l'identificazione del tipo e dell'origine delle cellule leucemiche. I quadri clinico ed ematologico di queste forme sono quelli classici delle LA.
i) Leucemia congenita.
Come leucemia acuta congenita, di cui si conoscono più di 100 casi, si intende una LA che si manifesti entro la prima settimana di vita (v. Wintrobe, 19747, p. 1476). La stragrande maggioranza di detti casi, come avviene per le LA insorgenti nel primo anno di vita, è di tipo mieloide, contrariamente a quanto avviene nei bambini e nei giovani in cui vi è netta preponderanza di LLA. Caratteri della LA congenita sono la mancanza di anemizzazione, la presenza di infiltrati cutanei e la scarsissima sensibilità alla terapia.
l) Sindrome preleucemica o dismielopoietica o mielodisplastica.
È questo uno dei capitoli più tormentati di tutta l'ematologia. Anzitutto occorrerebbe eliminare la confusione terminologica derivante dalla copiosa serie di sinonimi con cui la sindrome è indicata in letteratura: leucemia oligoblastica o pauciblastica, leucemia subacuta, anemia refrattaria con eccesso di blasti, smouldering leukemia (v. Straus e altri, 1980). Alcuni autori, per motivi di ordine e razionalità, sotto il termine di sindromi mielodisplastiche acquisite hanno raggruppato varie situazioni patologiche: la concezione fondamentale di C. Sultan (v., 1977) è riportata nella tab. XI, in cui è stato aggiunto il sarcoma granulocitico che talvolta può precorrere l'insorgenza della LA (v. Mason e altri, 1973).

Volendo semplificare al massimo, si può dire che esistono condizioni clinico-ematologiche particolari, estrinsecantisi perifericamente o solo con anemia di tipo normo- o macrocitico, oppure con pancitopenia, cui corrisponde un reperto midollare senza segni di LA, che entro intervalli di tempo variabili ‛possono, ma non debbono', evolvere in LA.
Esistono ancora altre situazioni, caratterizzate da quadri periferici simili a quelli precedentemente ricordati e dalla presenza nel midollo di un eccesso di mieloblasti e promielociti mostranti talvolta atipie morfologiche, che in una certa percentuale evolveranno in LA. Le LA che originano in senso lato da sindromi preleucemiche sono invariabilmente di tipo mieloide più o meno differenziato.
m) Leucemie miste.
Si designano con questo termine situazioni patologiche in cui, nello stesso paziente, si verifica l'insorgenza di un processo eteroplastico mieloproliferativo successivamente a uno linfoproliferativo: per esempio in un soggetto affetto da LLC subentra una LMC oppure una LMA. Sono noti anche rari casi in cui alla LLA è seguita, durante la fase di remissione completa, una LMC (v. Mauri e altri, 1977).
n) Leucemie mieloidi croniche giovanili.
Sono indubbiamente rare e rappresentano solo dall'1,5 al 5% di numerose casistiche (v. Rizzo e altri, 1979). Se ne distinguono due forme: una perfettamente sovrapponibile a quella dell'adulto, Ph1 positiva; l'altra Ph1 negativa, identificata solo da meno di vent'anni, che si manifesta in soggetti molto giovani e ha caratteri clinici di evoluzione e risposta terapeutica particolari (v. Galton, 1974).
o) Leucemia mieloide cronica senza cromosoma Filadelfia.
Esistono rari casi di LMC in cui, anche usando le tecniche più moderne, non è dimostrabile il cromosoma Ph1 (v. i contributi di Rowley, 1978): essi si distinguono per l'età piuttosto avanzata dei soggetti, per la scarsa sensibilità alla terapia classica della LMC e per la sopravvivenza più breve.
p) Leucemia mielomonocitica cronica.
Forma ancora non perfettamente identificata, che presenta però caratteri clinici ed ematologici abbastanza precisi (v. Geary e altri, 1975; v. Rizzo e altri, 1979), in passato e anche attualmente confusa in genere con forme atipiche di LMC in cui è possibile il reperto di monocitosi. Questo tipo di leucemia può insorgere anche in bambini e giovinetti e rientra nel gruppo dei casi descritti come LMC giovanile Ph1 negativa.
q) Leucemia monocitica cronica.
Molto rara, colpisce in genere soggetti anziani ed è ematologicamente caratterizzata da leucocitosi sostenuta dal forte aumento di monociti circolanti. L'andamento della malattia è in genere benigno e non necessita pertanto di terapia.
r) Leucemia neutrofila cronica.
Questa malattia, eccezionalmente rara ma la cui esistenza è ammessa dai più autorevoli trattatisti (v. Galton, 1974), colpisce soggetti anziani ed è caratterizzata ematologicamente da leucocitosi per aumento dei neutrofili e clinicamente dalla splenomegalia e dal decorso cronico benigno non richiedente terapia.
s) Leucemia basofila cronica e leucemia eosinofila cronica.
La reale esistenza di questi due tipi di leucemie croniche è ancora oggetto di discussione.
t) Tricoleucemia o hairy cell leukemia.
Negli ultimi anni molto si è discusso su questo tipo di leucemia (v. Storti, 1981), caratterizzata clinicamente soprattutto da splenomegalia e dall'andamento cronico ma progressivo, ed ematologicamente in genere da pancitopenia di diverso grado. Il tipico rilievo morfologico - particolarmente evidente all'osservazione con il microscopio a contrasto di fase - di spicule o capelli delle cellule leucemiche del sangue periferico (v. fig. 19) ha valso a questa forma il termine di tricoleucemia.
6. Diagnosi e prognosi.
a) Premessa generale ed elementi di terminologia.
Il decorso o storia naturale della malattia leucemica mostra profonde differenze tra forme acute e forme croniche. I dati relativi all'era preterapeutica forniscono precise indicazioni sulla sopravvivenza a tale malattia se non trattata: 3-4 mesi per le LA, 28 mesi per la LMC, 4-6 anni per la LLC. Le varie e complesse possibilità terapeutiche, cioè sia quelle per la cura della malattia e delle sue complicanze sia quelle di supporto, ne hanno ora modificato profondamente il decorso, soprattutto per quanto riguarda le LA, che possono presentare un succedersi di remissioni e di recidive. Di regola le successive remissioni sono sempre più brevi e le recidive di sempre più difficile controllo.
Dalla complessità del problema, dalle svariate risorse terapeutiche a nostra disposizione e dai risultati indubbiamente brillanti che ne conseguono, fino alla guarigione, sorge l'imperativo categorico di non affrontare mai queste malattie con fatalismo e rassegnazione: tale passivo atteggiamento appartiene fortunatamente a un'altra epoca, mentre, se gli attuali risultati sono già confortanti, la grande mole di ricerche compiute e le forze nuove che continuamente vi si dedicano sul piano sperimentale inducono a riporre una fondata fiducia nella possibilità di chiarire in un prossimo futuro i punti ancora irrisolti. Per una migliore comprensione di quanto segue appare utile illustrare la terminologia di uso corrente in terapia ematologica.
La ‛remissione completa' (RC) è una condizione la cui diagnosi può essere posta solo in base allo stato del midollo osseo. Gli elementi che consentono di ritenere che un soggetto affetto da leucemia è in RC sono: assenza di segni clinici imputabili alla malattia, normalità dell'esame emocromocitometrico, quota dei blasti leucemici presenti nel midollo osseo non superiore al 5%. Secondo vari autori, e noi tra questi, in un soggetto in RC non si dovrebbero osservare cellule leucemiche nel midollo. È doveroso ricordare che la remissione completa è il traguardo della terapia della LA, perché è ormai fuori discussione che uno dei fattori prognostici più importanti per la sopravvivenza è la durata della prima RC (v. Rizzo e altri, 1980).
Si parla di ‛remissione parziale' (RP) quando la condizione risultante a seguito della terapia non è caratterizzata dai parametri ematologici che definiscono la RC.
Si parla di ‛recidiva' quando compaiono manifestazioni cliniche e/o ematologiche imputabili a LA dopo che il paziente ha beneficiato di un periodo variabile di RC o RP. Le recidive possono essere midollari oppure extramidollari e perciò variamente localizzate (testicolo, meningi, ovaio, ecc.): per i due tipi prognosi e terapia sono diverse.
b) Leucemie acute.
La diagnosi di LA è in genere facile per l'evidenza sia dei dati clinici sia soprattutto di quelli ematologici. Solo i casi in cui manchi la leucocitosi o le alterazioni cellulari siano poco rilevanti, oppure quelli nei quali, malgrado la presenza di certi quadri clinici, si trascura di procedere ad accertamenti ematologici banali, quali l'emogramma e la formula leucocitaria, possono indurre in errore il medico non specialista o poco esperto. I capisaldi della diagnostica ematologica sono: leucocitosi sostenuta dalla presenza in circolo di cellule atipiche (biasti leucemici), infiltrazione midollare di grado diverso e fino alla completa sostituzione da parte di tali blasti, monomorfismo cellulare periferico e midollare, hiatus leucemicus. Esistono indubbiamente situazioni ematocliniche che costituiscono difficoltà diagnostiche anche per sperimentati ematologi e altre invece che possono essere correttamente interpretate, come abbiamo già esposto, mediante l'impiego di tecniche particolari: a tale proposito, sembra opportuno mettere ancora in evidenza l'importanza che nella diagnostica di malattie così gravi assume l'adozione di semplici accorgimenti tecnici.
Per quanto riguarda la prognosi occorre anzitutto operare una distinzione tra leucemie non linfoblastiche e LA linfoblastica, e di questa un'ulteriore suddivisione in LLA dei bambini e dei giovani e LLA degli adulti, perché nelle varie forme sono diversi i risultati terapeutici nei riguardi sia della possibilità di indurre remissione completa sia della durata della sopravvivenza. È noto infatti che la possibilità di ottenere RC nei bambini supera il 90% dei casi mentre negli adulti è nettamente inferiore, anche se per gruppi selezionati di pazienti adulti sono state riportate percentuali simili (v. Rizzo e altri, 1980). Anche la durata della prima RC e quella della sopravvivenza sono significativamente più lunghe nei bambini che negli adulti: da uno studio cooperativo condotto in Italia (v. A.I.L., 1979) è risultato che il 56% dei soggetti era ancora in RC dopo tre anni, mentre altri ricercatori hanno fissato al 70% la probabilità che i bambini sopravvivano per 5 anni e al 40% la probabilità per gli adulti di sopravvivere per 4 anni. È infatti specialmente tra i bambini che si osserva un discreto numero, pari o superiore al 5% a seconda degli autori, di lunghe sopravvivenze, cioè oltre gli otto-dieci anni: sono questi i casi in cui si può ritenere che la leucemia sia guarita. Casi simili si possono osservare, seppure, purtroppo, assai raramente, anche tra gli adulti: un nostro paziente, che aveva 35 anni al momento dell'insorgenza della LLA, si è sposato durante il ricovero in ospedale, ha avuto due figli e da oltre 10 anni è in ininterrotta prima RC.
Appare ovvia l'importanza di acquisire elementi di giudizio che, al momento della diagnosi, consentano non solo di formulare la prognosi sulla probabile evoluzione della malattia, ma anche di modulare la terapia. A tale argomento è stato dedicato un simposio internazionale (v. Fliedner e Perry, 1975) e numerosissimi lavori (v., per la letteratura, A.I.L., 1979; v. Rizzo e altri, 1980). Mentre non esistono dubbi circa l'importanza di alcuni di tali elementi, per altri il significato non è univoco; per quanto riguarda la LLA, essi sono riassunti nella tab. XII.

Nelle LA non linfoblastiche, che sono un coacervo di vari tipi (v. tab. I), i risultati ottenuti per quanto riguarda sia la percentuale di RC indotte sia la sopravvivenza non sono così brillanti come nella LLA. È importante notare che non esistono sostanziali differenze tra bambini e adulti. Attualmente le RC sono indotte in circa il 50% dei casi (v. Mertelsman e altri, 1980), anche se da alcuni centri ospedalieri sono state riportate percentuali maggiori, fino al 70-80%: ma tali risultati non sono stati ottenuti da altri, pur usando gli stessi schemi terapeutici. La sopravvivenza dei pazienti trattati con la sola polichemioterapia è generalmente compresa tra 6 e 21 mesi, più lunga quando vi si associ l'immunoterapia.
Ammesso che tra i diversi tipi di LA non linfoblastiche vi sia una certa differenza - limitata tuttavia a pochi mesi - sia nella percentuale delle RC ottenibili sia nella sopravvivenza (ibid.), la presenza dei bastoncini di Auer rappresenterebbe l'unico elemento prognostico valido di significato più favorevole, secondo i dati di due ampie casistiche (v. Henderson e altri, 1975; v. Mertelsman e altri, 1980).
c) Leucemie croniche.
Come abbiamo già detto, per le LMC esistono due fasi ben distinte: 1) una fase cronica che, nella maggior parte dei casi, sfocia in 2) una fase accelerata esitante in genere nella crisi blastica. È indubbio che con gli attuali trattamenti si possono indurre remissioni della sintomatologia clinica e anche ematologica, ma non la RC, perché, per esempio, i tentativi tesi a eradicare il clone cellulare Ph1+ sono risultati negativi. D'altro canto è noto, da osservazioni compiute nell'era preterapeutica, che nella fase cronica la malattia può presentare periodi di miglioramento spontaneo, cioè remissione parziale spontanea. La diagnosi della LMC nella forma tipica non presenta problemi di sorta: al rilievo clinico di splenomegalia corrisponde ematologicamente, nel sangue periferico, leucocitosi con presenza nella formula leucocitaria di elementi immaturi della serie mieloide, senza salti maturativi. Sono in genere presenti in circolo metamielociti e mielociti, qualche mieloblasto e promielocito, e frequentemente sono aumentati i basofili e gli eosinofili circolanti. La mielobiopsia conferma l'iperplasia granuloblastica e gli altri aspetti del midollo osseo già esposti nel cap. 4. Lo score della fosfatasi alcalina leucocitaria è molto basso. Nei casi dubbi la diagnosi si basa sulla dimostrazione della presenza del cromosoma Ph1 nel cariogramma (v. fig. 4).
Per quanto attiene alla sopravvivenza, i miglioramenti ottenuti rispetto all'era preterapeutica e a quella in cui l'unico trattamento possibile era il radiante non sono stati brillantissimi: infatti si è passati da una sopravvivenza media di 31 mesi (v. Minot e altri, 1924) a quella attuale di circa 40 mesi. Dalle casistiche si evince però che l'arco di sopravvivenza spazia da 1 a più di 10 anni, il che tra l'altro spiega come in gruppi particolari di soggetti la sopravvivenza media sia nettamente superiore a quella calcolata nella totalità dei pazienti (v. Rizzo e altri, 1979). Una volta che si è instaurata la crisi blastica, le possibilità di indurre RC sono scarse, a meno che il clone cellulare blastizzante non sia di tipo linfoide: in tal caso instaurando la chemioterapia usata per la LLA si induce RC in un'alta percentuale di casi. La sopravvivenza alla crisi blastica è di pochi mesi: nel caso della crisi di tipo linfoide raggiunge, al massimo, quella che si osserva nell'adulto affetto da LLA.
La LLC è un'affezione con un vasto spettro di presentazioni cliniche ed ematologiche: accanto a casi con andamento relativamente aggressivo, in cui la sopravvivenza è solo di qualche anno, ve ne sono altri caratterizzati da un comportamento biologico più blando e da una sopravvivenza di 15-20 anni e più. Va ascritto a Rai e altri (v., 1975) il merito di aver proposto una classificazione clinica della LLC in vari stadi, a ciascuno dei quali corrisponde una diversa sopravvivenza (v. tab. V), classificazione che ha ricevuto successivamente ampia conferma (v. Galton, 1977).
La diagnosi della LLC non presenta difficoltà: nella forma tipica il soggetto che ne è affetto presenta adenomegalie superficiali simmetriche e non dolenti, adenomegalie profonde, talvolta splenomegalia e, dal punto di vista ematologico, leucocitosi periferica in genere moderata, con netta linfocitosi assoluta e infiltrazione midollare linfocitaria di almeno il 30-40%, sostenuta da linfociti piccoli, maturi.
Nei casi con scarsa o assente leucocitosi si può ricorrere alla determinazione delle immunoglobuline di superficie dei linfociti: se queste sono di tipo monoclonale è stabilita la natura leucemica della malattia.
L'exitus è in genere da ascriversi a complicanze infettive, la cui frequenza è in rapporto al progressivo deteriorarsi della reattività immunologica del paziente, o a fenomeni emorragici legati alla progressiva piastrinopenia, oppure a un'inarrestabile cachessia. Rarissima è la comparsa di una crisi blastica terminale, caratterizzata ematologicamente dalla presenza e prevalenza in circolo di linfoblasti.
7. Complicanze.
Le complicanze sono abbastanza frequenti durante il decorso non solo delle LA, ma anche delle leucemie croniche, specialmente della LLC. Le ricorderemo succintamente.
Nel corso delle LA le complicanze più frequenti nella fase di acuzie sono quelle infettive ed emorragiche, con nettissima prevalenza delle prime. In tale fase alcune complicanze sono di frequente riscontro in determinati tipi di LA, come la CID nella LA promielocitica. Altre possono manifestarsi nella fase di induzione della RC ed essere in rapporto all'aplasia midollare provocata dalla terapia citostatica, o avere comunque origine iatrogena essendo causate dalle varie terapie praticate, oppure - anche se molto più raramente - possono rappresentare una conseguenza dell'evoluzione della malattia di base. Complicanze di vario tipo e causa possono presentarsi anche durante la fase di mantenimento; le più frequenti sono indubbiamente quelle di tipo infettivo, sia batterico sia virale, favorite tra l'altro dall'azione dei farmaci citostatici usati per la terapia di mantenimento, specie se nell'ambito di schemi aggressivi, farmaci che com'è noto sono tutti immunodepressori. Complicanze di più rara osservazione in tale fase sono le pneumopatie, l'insufficienza epatica, le localizzazioni cerebrali. Possibili e numerose sono le complicanze di natura iatrogena per l'azione tossica generica o specifica dei citostatici (v. Weiss e altri, 1974; v. Ghione, 1977; v. Mauer, 1980; v. tab. XIII).

Nella LMC le complicanze reali nella fase cronica, se pure esistono, sono rarissime: tra queste possiamo ricordare il priapismo e le trombosi venose. In genere nel passato erano ascritte a tale fase varie complicanze che in realtà appartengono alla fase accelerata, acuta, e ricalcano quelle già descritte per la LA. La complicanza più frequente è di tipo iatrogeno, indotta dal farmaco maggiormente usato per la cura della LMC, il busulfano (v. tab. XIII). E noto che il suo uso prolungato può indurre fibrosi polmonare, il cosiddetto polmone da busulfano, oltre a piastrinopenia, aplasia midollare, pigmentazione scura (melaninica) della cute, amenorrea, oligospermia e/o azoospermia. Anche un altro farmaco che si usa nella LMC, il dibromomannitolo, può indurre fibrosi polmonare, amenorrea e pigmentazione cutanea.
Il decorso della LLC è segnato dall'alta frequenza delle complicanze infettive, sia ripetute, sia episodiche, che colpiscono più del 50% dei soggetti (v. Miller, 1962), perché la LLC si accompagna ad alterazioni della reattività immunologica e in particolare della produzione delle immunoglobuline; molto più raramente è implicata la neutropenia. Frequente è anche l'insufficienza midollare che determina citopenia periferica: anemia, neutropenia, piastrinopenia, isolate o variamente combinate fino alla pancitopenia. Un altro gruppo di complicanze relativamente frequenti sono quelle di ordine autoimmunitario, in primo luogo l'anemia emolitica autoimmune che complica circa il 15-20% delle LLC e che può insorgere in qualunque momento del decorso della malattia; può talvolta osservarsi anche una piastrinopenia a eziologia autoimmune.
L'insorgenza di seconde neoplasie sembra osservarsi con maggiore frequenza in pazienti affetti da LLC rispetto a un'equipollente popolazione (v. Galton, 1977): la più nota e frequente di tali associazioni è rappresentata dalla sindrome di Richter, caratterizzata dalla comparsa di sarcoma linfonodale in corso di LLC.
8. Terapia.
La terapia delle leucemie è un argomento che si è sviluppato, specialmente per quanto concerne la LA, negli ultimi trentacinque anni. Prima di allora solo per le leucemie croniche si disponeva di possibilità terapeutiche, rappresentate dalla soluzione arsenicale di Fowler, medicamento introdotto da Lissauer (v., 1865) per il trattamento della LMC, e dalle radiazioni X, proposte da Pusey (v., 1902) e usate sia per la LMC sia per la LLC (v. Storti, 1978, per la storia della terapia).
a) Concetti attuali di terapia delle leucemie acute.
Dall'introduzione in terapia, nel 1948, del primo farmaco antileucemico, l'amminopterina antagonizzante l'acido folico, le possibilità terapeutiche si sono enormemente espanse.

Oggi l'elenco dei farmaci a provata azione antileucemica è nutrito (v. tab. XIV) e di essi si conoscono bene le modalità d'azione (v. fig. 7). Inoltre è stato chiarito che tali farmaci esplicano il massimo dell'azione terapeutica quando siano impiegati secondo determinati schemi polichemioterapici, e sono stati messi a punto schemi terapeutici praticamente specifici per i diversi tipi di LA: ne consegue pertanto la necessità, in ogni caso di LA, di compiere ogni sforzo per determinarne l'esatta classificazione. È indispensabile poi tentare di indurre RC, in quanto solo ottenendo tale risultato si può sperare di sfruttare al massimo l'attuale terapia antileucemica. Una eventuale RP dovrà rappresentare un risultato non voluto e ricercato, ma solo inevitabilmente accettato.
Attualmente la sequenza terapeutica nel trattamento delle LA comprende: 1) induzione della RC; 2) consolidamento della RC; 3) profilassi delle possibili localizzazioni extramidollari; 4) terapia di mantenimento; 5) altre terapie; 6) sospensione di ogni terapia.
Va chiarito che i notevoli risultati terapeutici che si possono attualmente vantare sono la conseguenza di una pura ricerca empirica clinica, in quanto non conoscendosi l'eziopatogenesi della LA non è possibile instaurare una terapia sicuramente eziologica. Dobbiamo ancora sottolineare che la condotta terapeutica ha mirato per un lungo periodo alla completa eliminazione delle cellule leucemiche dall'organismo; successivamente gli studi di citocinetica hanno dimostrato l'irraggiungibilità di tale traguardo (v. Gavosto, 1977; v. Gunz e Vincent, 1977) e anche la ricerca sperimentale lo ha confermato, dimostrando che si selezionano cellule leucemiche resistenti (v. Skipper e altri, 1978). Attualmente si tenta di diminuire la massa di cellule leucemiche dell'organismo affinché il sistema di sorveglianza immunitario riprenda il sopravvento, ed è possibile spingere la terapia a livelli prima impensabili e controllare molte complicanze del decorso grazie alla cosiddetta ‛terapia di supporto' che ha raggiunto mete solo pochi anni fa insperate (v. Bodey, 1976; v. Lister e Yankee, 1978).
b) Induzione della remissione completa.
Scopo di questa prima fase è di ottenere rapidamente la riduzione del numero dei blasti leucemici, impiegando dosi di farmaci che causino la più bassa tossicità, al fine di permettere il ripristino degli elementi midollari normali. Si ritiene che al momento della diagnosi siano presenti nell'organismo 1012 cellule leucemiche e che al termine dell'induzione, quando il paziente è in RC (in cui praticamente non sono reperibili blasti leucemici), il loro numero sia ridotto a 109-1010. La fase di induzione dura tre-quattro settimane e il rapido raggiungimento della RC, che come si è detto è un fattore essenziale per sperare di controllare a distanza di tempo la malattia, costituisce un importante segno prognostico favorevole.
c) Consolidamento della remissione completa.
Ottenuta la RC si può tentare di ridurre ulteriormente la massa cellulare leucemica con un periodo breve di intensa polichemioterapia. Questo momento della strategia terapeutica non è però obbligatorio né applicato in tutti gli schemi terapeutici, perché, tra l'altro, comporta il rischio di un aumento delle morti in RC, specie per infezioni, in conseguenza dell'azione immunosoppressiva dei citostatici.
d) Profilassi delle possibili localizzazioni extramidollari.
È questa una fase di primaria importanza nella terapia della LLA, specie dei bambini, e riguarda essenzialmente il trattamento delle localizzazioni nel sistema nervoso centrale; è stata impostata e schematizzata dal gruppo del St. Jude Children's Research Hospital nel programma di ‛terapia totale' teso all'eradicazione della malattia leucemica (v. Simone, 1974; v. Hustu e Aur, 1978).
L'intervento terapeutico in questa fase dovrà anche essere rivolto a eradicare possibili localizzazioni nel testicolo, che al pari del SNC costituisce, come s'è già detto, un ‛santuario' ove i blasti leucemici possono allogarsi senza essere raggiunti dai citostatici somministrati per via generale (v. Hustu e Aur, 1978).
e) Mantenimento.
Allorché è indotta la RC, se non si procede all'ulteriore somministrazione di citostatici la recidiva interviene entro breve tempo. La terapia di mantenimento, basata in genere sull'impiego di più farmaci, viene pertanto instaurata al fine di mantenere il più a lungo possibile la RC.
f) Altre terapie.
Nell'ambito della terapia di mantenimento è attuabile la reinduzione ciclica, mediante la somministrazione a determinati intervalli di tempo degli stessi farmaci che indussero la remissione completa o di altri citostatici.
Per migliorare i risultati terapeutici è possibile usare l'immunoterapia, che però è ancora oggetto di studio e di dibattito (v. Alexander e Powles, 1978; v. Whittaker, 1980), cosi come ancora a livello di studio e sperimentazione dev'essere considerato il trapianto di midollo nel trattamento della LA (v. Sanders e Thomas, 1978).
L'immunoterapia è distinta in attiva, passiva e adottiva. L'immunoterapia attiva, basata sulla stimolazione delle difese dell'ospite contro la malattia, può essere specifica o aspecifica; la prima è realizzata mediante iniezioni settimanali di un pool di cellule leucemiche, provenienti da diversi malati con lo stesso tipo di leucemia e pretrattate con irradiazioni o con farmaci; la seconda è realizzata con somministrazioni settimanali, attraverso scarificazioni della cute, di vaccini aspecifici tipo BCG, o di Corynebacterium parvum o di Bordella pertussis. I due tipi di immunoterapia attiva possono essere impiegati separatamente o contemporaneamente. L'immunoterapia passiva consiste invece nella somministrazione di sieri o di immunoglobuline di donatori immunizzati. L'immunoterapia adottiva si basa sul trasferimento nel paziente di cellule immunocompetenti, provenienti da soggetti normali o da leucemici in remissione: consiste quindi, in pratica, in un trapianto di midollo osseo omologo.
Ripetiamo che l'argomento, che impegna da svariati anni eminenti ricercatori, può attualmente essere solo trattato come a sé stante: se ne sottolineeranno di volta in volta i riflessi nell'ambito della terapia delle LA, così come verrà fatto per il trapianto di midollo.
g) Sospensione della terapia.
Come si è già accennato, i citostatici esercitano effetti tossici a vari livelli (v. Weiss e altri, 1974; v. Ghione, 1977; v. Mauri, 19803; v. tab. XIII; v. chemioterapia antineoplastica) dei quali in particolare sono sconosciuti gli sviluppi nel tempo. Agli ematologi si prospetta quindi il problema della scelta del momento in cui sospendere ogni terapia in pazienti da lungo tempo in RC: attualmente tale limite è posto a 24-36 mesi di ininterrotta RC, ma secondo chi scrive è conveniente prolungare la terapia per almeno 36 mesi e ancora oltre (48-60 mesi) in quei casi che presentino segni prognostici particolarmente sfavorevoli.
h) Caratteristiche generali della chemioterapia.
La RC può essere ottenuta usando un solo farmaco, cioè instaurando una monochemioterapia, oppure utilizzando più farmaci, cioè una polichemioterapia standardizzata secondo schemi precisi, con la quale si ottiene una percentuale di RC nettamente più alta, e che pertanto dev'essere preferita. Tra i vari citostatici antileucemici non esiste equipollenza terapeutica nelle varie fasi della malattia; pertanto si suole distinguere tra citostatici utili per la fase di induzione e per la fase di mantenimento. I citostatici esercitano, alcuni inevitabilmente, severi effetti tossici collaterali (v. tab. XIII) e posseggono sempre azione immunodepressiva, donde la necessità che la terapia della LA sia prescritta e condotta da specialisti di sicura esperienza.
Tratteremo ora più specificamente delle modalità terapeutiche riguardanti le due forme paradigmatiche di LA, la LLA e la LMA, limitandoci però a illustrare i presidi essenziali e a fornire le indicazioni bibliografiche più recenti, utili per l'ulteriore approfondimento dell'argomento.
i) Leucemia linfoblastica acuta.
Allorché sia posta diagnosi di LLA si deve iniziare al più presto la terapia, dopo aver controllato che il paziente non presenti situazioni che ne controindichino l'inizio, quali stati infettivi e/o sindromi emorragiche o coagulopatie gravi, che dovranno essere opportunamente trattati (per la letteratura più recente, v. Bodey e Rodriguez, 1978; v. Mauer, 1980; v. Mauri, 19803; v. Rizzo e altri, 1980; v. Simone, 1980). Gli schemi terapeutici proposti e usati nel tempo sono diversi (v. Bodey e Rodriguez, 1978), ma oggi si ritiene unanimemente che l'associazione vincristina + prednisolone determini percentuali di RC non superate da altri schemi polichemioterapici. Si suole però associare un altro citostatico, un antibiotico antraciclinico quale la daunorubicina o l'adriamicina, oppure un enzima, la L-asparaginasi, in quanto l'adozione di tale schema, pur non essendo seguita da un aumento della percentuale di RC, consente di aumentare la percentuale dei pazienti che beneficeranno di una ininterrotta prima remissione (v. Mauer, 1980). L'esatto schema terapeutico, completo delle indicazioni posologiche, è il seguente: vincristina, 2 mg/m2 di superficie corporea per via endovenosa 1 volta la settimana, per 4-6 somministrazioni; daunorubicina, 60 mg/m2 di superficie corporea per via endovenosa 1 volta la settimana (in genere 48 ore dopo la vincristina) per 4-6 somministrazioni; prednisolone, 60-120 mg/m2 di superficie corporea per os al giorno, per 4-6 settimane.
Nei soggetti di oltre 60 anni, all'antibiotico si può sostituire la L-asparaginasi, perché è purtroppo noto che i citostatici antraciclinici sono particolarmente cardiotossici (v. Ghione, 1977; v. tab. XIII).
Allorché si è indotta la RC, si deve subito procedere, specialmente nei bambini, alla profilassi delle recidive a livello del sistema nervoso centrale: è stato infatti dimostrato che in circa il 50% dei bambini in RC tale distretto è la sede della prima e relativamente precoce recidiva (v. Simone, 1974; v. Fontana e altri, 1975; v. Fontana e Rizzo, 1976; v. Mauer, 1980). La necessità di disporre di un sistema idoneo a evitare questo gravissimo ostacolo al mantenimento ininterrotto della RC ha dato origine al seguente schema: irradiazione del cranio con 2.400 rad+methotrexate (10 mg/m2 di superficie corporea) per via endorachidea per 4 volte.
Tale profilassi ha ridotto enormemente il pericolo delle localizzazioni encefaliche (v. Fontana e altri, 1975; v. Fontana e Rizzo, 1976), ma la possibilità che sia seguita nel tempo da tutta una serie di effetti secondari indesiderabili (v. Mauer, 1980) ha indotto a ricercare il minimo dosaggio di irradiazione caratterizzato dallo stesso elevato indice terapeutico (v. Mauer, 1980; v. Simone, 1980).
La successiva fase terapeutica di mantenimento è basata sull'impiego di due altri citostatici, la 6-mercaptopurina e il methotrexate, generalmente secondo il seguente schema: 6-mercaptopurina, 40-60 mg/m2 di superficie corporea per os al giorno, + methotrexate, 7-10 mg/m2 di superficie corporea per via intramuscolare 2 volte la settimana.
Su questo punto le opinioni dei clinici non sono concordi, perché alcuni (v. Mauer, 1980) proseguono ininterrottamente tale schema di mantenimento fino alla sospensione di ogni terapia o alla comparsa di una recidiva; altri invece, e noi tra questi, procedono regolarmente a nuove somministrazioni dei farmaci sia per via generale sia per via endorachidea.
Non sembra che nella LLA l'immunoterapia consenta di ottenere risultati migliori rispetto alla chemioterapia (v. Alexander e Powles, 1978; v. Mauer, 1980; v. Whittaker, Immunotherapy..., 1980).
l) Leucemia mieloide acuta.
Sebbene il trattamento di questo tipo di LA si basi sugli stessi concetti generali che ispirano la condotta terapeutica nella LLA, rispetto a quest'ultima si ottengono, sia come percentuale e durata della RC, sia come sopravvivenza, risultati senz'altro meno brillanti, per cui attualmente si è ancora alla ricerca dello schema terapeutico più appropriato (v. Bodey e Rodriguez, 1978; v. Wiernik, 1978; v. Gale, 1979; v. Mauri, 19803; v. Whittaker, Advances in..., 1980). Un netto aumento della percentuale di RC ha fatto seguito all'introduzione in terapia dell'arabinoside citosina e della daunorubicina impiegate secondo vari schemi polichemioterapici (v. Bodey e Rodriguez, 1978; v. Wiernik, 1978; v. Whittaker, Advances in..., 1980) dei quali riportiamo qui quello da noi adottato da anni con ottimi risultati (v. Rees e altri, 1977): daunorubicina, 50 mg/m2 di superficie corporea per via endovenosa al primo giorno di trattamento; arabinoside citosina, 100 mg/m2 di superficie corporea per via endovenosa ogni 12 ore dal 1° al 5° giorno di trattamento; tioguanina, 100 mg/m2 di superficie corporea per os ogni 12 ore dal 1° al 5° giorno di trattamento.
In considerazione dei risultati complessivamente deludenti della terapia mirante a indurre la prima RC, non è ancora possibile indicare uno schema unico di terapia di mantenimento, per il quale sono tuttora in corso ricerche. Se si eccettuano schemi che sono ancora in fase di valutazione (v. Wiernik, 1978; v. Whittaker, Advances in..., 1980), attualmente la condotta terapeutica maggiormente seguita consiste nel somministrare, a regolari intervalli di tempo, gli stessi farmaci usati per l'induzione, oppure nell'attenersi ad altri schemi terapeutici che escludano i citostatici antraciclinici per evitarne la cardiotossicità.
Le ricerche condotte negli ultimi anni sul possibile impiego dell'immunoterapia nel trattamento della LMA hanno consentito di dimostrare che i risultati che ne conseguono non sono brillanti e che il suo effetto principale consiste nel determinare un certo allungamento della sopravvivenza, in quanto in pazienti così trattati sono più facilmente inducibili nuove RC e comunque remissioni parziali dopo la prima e anche dopo la seconda recidiva (v. Alexander e Powles, 1978; v. Wiernik, 1978; v. Whittaker, Immunotherapy..., 1980). Si ricorda che solo in alcuni centri altamente specializzati si eseguono due altre pratiche terapeutiche, il trapianto di midollo allogenico, cioè di gemello monocoriale ovverosia HLA identico, e il trapianto autologo, cioè il trapianto di midollo dello stesso paziente prelevato durante RC e crioconservato (v. trapianti), terapie sulla cui validità non sono ancora disponibili dati sicuri.
m) Terapia di supporto.
Con questo termine si designa la serie di interventi terapeutici che, atti a fronteggiare e risolvere le complicanze che possono insorgere specialmente nelle prime fasi del decorso della LA, contribuiscono al conseguimento dei brillanti risultati nell'attuale trattamento di questa affezione. Così, per fronteggiare le complicanze infettive risultano preziosi soprattutto i nuovi antibiotici che continuamente vengono prodotti, la possibilità di trasfondere granulociti ottenuti mediante le tecniche di separazione cellulare, e infine la pratica della degenza nelle cosiddette ‛camere sterili' per ridurre drasticamente i pericoli di contagio; per sopperire all'anemizzazione e alla piastrinopenia, causa di diatesi emorragica, validi e insostituibili sussidi terapeutici sono la somministrazione di concentrati eritrocitari e, rispettivamente, di elevatissime quantità di piastrine, sempre ottenuti mediante il separatore cellulare. Ancora, è possibile, con appropriati trattamenti, fronteggiare il pericolo delle coagulopatie, di cui la più frequente è la CID, mentre sono in corso di sperimentazione, e sottoposti al vaglio della critica, altri mezzi volti a ridurre l'incidenza delle più frequenti complicanze.
n) Terapia delle recidive.
L'insorgenza della recidiva, che fatalmente si verifica in numerosi pazienti affetti da LA e che purtroppo costituisce il segno prognostico peggiore, non deve esimere il medico dall'obbligo di compiere il massimo sforzo terapeutico per ottenere una nuova RC, in modo particolare quando i malati siano bambini: specie in questi casi, infatti, la seconda RC puù essere lunghissima, tanto da far ritenere, per il tempo trascorso dalla recidiva, che i pazienti siano guariti, come risulta dai sia pur rari casi riferiti dalla letteratura. Dal punto di vista biologico, l'insorgenza della recidiva significa che l'insieme delle terapie eseguite non è stato sufficiente a prevenire e controllare la riespansione della popolazione leucemica. I fattori che possono determinare tale fenomeno sono: 1) la farmacoresistenza; 2) l'interessamento da parte della malattia di organi non raggiungibili dai citostatici somministrati per via generale.
La recidiva può interessare il midollo osseo oppure altre sedi - meningi, encefalo, testicoli, ovaio, occhio - prospettando problemi di prognosi e di terapia differenti da caso a caso. La percentuale delle recidive nei vari organi in corso di LLA è riportata nella tab. XV. Le recidive nella LMA, di cui sono rarissime quelle extramidollari, presentano un quadro clinico-ematologico simile a quello iniziale.

o) Terapia delle leucemie croniche.
Leucemia mieloide cronica. - È ormai ampiamente dimostrato che nel trattamento della LMC la chemioterapia si è rivelata nettamente superiore all'irradiazione della milza che, prima dell'introduzione in terapia dei citostatici, rappresentava la sola arma disponibile per combattere la malattia. L'efficacia della moderna chemioterapia non implica, però, la necessità di iniziare il trattamento in ogni caso non appena venga formulata la diagnosi di LMC, soprattutto se la malattia viene individuata del tutto accidentalmente in un soggetto che non presenti sintomatologia: in situazioni di questo tipo è conveniente sottoporre il paziente a controlli periodici e iniziare il trattamento solo quando il tipico quadro clinico-ematologico della LMC si sia chiaramente delineato.
Per il trattamento della LMC il farmaco di elezione è il busulfano, citostatico ad azione alchilante, ma si sono dimostrati efficaci, sia pure in minor grado, anche altri citostatici: il dibromomannitolo, l'idrossiurea e il melfalan. Lo schema terapeutico più seguito prevede la somministrazione di una dose standard giornaliera di busulfano, controllando settimanalmente il numero dei leucociti: infatti, il riscontro di un livello di leucociti di circa 100.000/mm3 impone la sospensione della terapia che potrà essere ripresa solo quando il loro numero torni al disopra dei 25.000/mm3. Durante ogni fase di terapia si dovrà associare alla somministrazione del citostatico quella di allopurinolo, un farmaco dotato della proprietà di bloccare la produzione dell'acido urico che, esaltata per l'enorme distruzione cellulare determinata dal trattamento antileucemico, potrebbe essere causa di gravi complicanze, quale la nefropatia uratica.
Gli altri farmaci possono essere vantaggiosamente impiegati quando si manifestino sindromi cliniche e/o ematologiche che controindichino l'uso del busulfano, il quale può di per se stesso dar luogo a complicanze. Ancora allo stato di sperimentazione nel trattamento delle LMC sono la splenectomia, l'immunoterapia e le leucoaferesi (v. Galton, 1977; v. Sokal, 1977).
La terapia della crisi blastica che può manifestarsi in corso di LMC è simile a quella della LA, in quanto prevede l'utilizzazione di schemi terapeutici rispettivamente per il trattamento della LLA, quando il clone blastizzante è linfoide, e della LMA quando il clone è mieloide (v. Canellos, 1977).
Leucemia linfatica cronica. - In questa forma, ancora più che nella LMC, è necessario valutare attentamente nel singolo paziente l'opportunità di instaurare la terapia (v. Galton, 1977; v. Wiltshow, 1977). In linea generale si può sostenere che in pazienti che presentino decorso molto lento, chiaramente cronico, si può procrastinare l'inizio della terapia, che va invece tempestivamente intrapresa allorché siano evidenti segni generali e/o ematologici di un certo significato.
Il citostatico più usato è il clorambucile, un farmaco alchilante anche questo, che va somministrato per os ogni giorno, alla dose di 0,1-0,2 mg/kg di peso corporeo, finché il numero dei leucociti scende fino a circa 10.000/mm3; buoni risultati si ottengono anche con la ciclofosfammide.
Nel trattamento delle LLC sono stati e sono tuttora utilmente impiegati anche gli adrenocorticosteroidi (cortisone e suoi derivati), responsabili tuttavia di non trascurabili effetti collaterali, tra cui quello proflogistico e proinfettivo, dovuti soprattutto alla loro azione immunodepressiva particolarmente temibile in questa malattia. Anche in questi casi durante ogni ciclo di terapia deve essere somministrato allopurinolo per evitare i danni renali derivanti dal sovraccarico di acido urico.
Trattamenti ancora in fase sperimentale sono l'irradiazione totale del corpo secondo tecniche particolari (v. Johnson, 1977) e la leucoaferesi (v. Galton, 1977).
È assolutamente necessario combattere nel modo più energico i processi infettivi, che rappresentano le più frequenti complicanze in corso di LLC, sia mediante l'uso di antibiotici a largo spettro e a piena dose, sia eventualmente con la somministrazione generosa di gammaglobuline per via parenterale. La possibile, sebbene meno frequente, comparsa di anemia emolitica autoimmune costituisce un'indicazione all'impiego degli immunosoppressori (adrenocorticosteroidi, ciclofosfammide, ecc.) e in qualche caso alla splenectomia.
9. Aspetti psicologici dell'assistenza al paziente leucemico.
A chi cura pazienti affetti da leucemia compete non solo la corretta applicazione della parte tecnica, ma anche il compito difficile e delicato dell'assistenza morale al malato e ai suoi familiari, compito che, assai più che in molte altre gravi malattie, richiede dedizione e pazienza. In tal senso, l'impegno richiesto al medico è ancora più profondo nei casi di LA, in particolare dei bambini. Un tempo la quasi totalità di questi casi decorreva così rapidamente e senza il benché minimo dubbio sull'esito irrimediabilmente fatale della malattia, che il compito dell'assistenza morale del medico non presentava eccessive difficoltà. L'attuale diffusione attraverso la stampa di notizie di sicure guarigioni di pazienti leucemici e la propaganda di tecniche terapeutiche d'avanguardia - quali l'immunoterapia, il trapianto di midollo osseo, l'uso del separatore cellulare - esaltano l'ansia dei familiari, che vedono sempre più frequentemente i piccoli malati in remissione completa tornare alla vita normale, alla scuola, persino allo sport, e sono da ciò spinti a porre ai sanitari pressanti e precisi quesiti.
Da molti anni, perciò, si dedica una sempre maggiore attenzione ai problemi psicologici del paziente e dei familiari (v. Baserga, 1972; v. Bahnson, 1975; v. Creech, 1975).
Per aver seguito, in quarant'anni di esperienza, centinaia di leucemici e migliaia di emoblastici in genere, siamo convinti che innanzi tutto, salvo casi eccezionali, sia sempre bene occultare al malato la verità; occorre comunque formulare una diagnosi che soddisfi il suo spirito critico di profano e offra alla sua mente un modello di processo morboso che preveda la possibilità di un decorso a poussées, lasciandogli però la speranza che, malgrado l'importanza della malattia, la prognosi sarà favorevole.
Pratica gravosa, ma di sicura utilità, è anche quella di parlare molto a lungo col malato e con i suoi parenti, perché così si riesce a migliorarne nettamente lo stato emotivo: il medico poco comunicativo aumenta l'ansia e lo sconforto dei familiari.
Infine, nel comunicare la diagnosi ai familiari come nel formulare la prognosi, occorrono da parte del medico cautela e delicatezza massime e un clima di profonda, umana solidarietà. Non si pronunci la parola ‛leucemia' e nel formulare la prognosi si abbia cura di usare grande equilibrio. Facendo presente la notevole gravità della malattia, si eviti tuttavia di cadere nell'errore di esprimere previsioni pessimistiche a brevissima distanza di tempo, in quanto il decorso della leucemia acuta nel caso singolo è assolutamente imprevedibile. La nostra casistica annovera leucemici vissuti per parecchi anni e alcuni da considerarsi guariti, per i quali al momento della diagnosi colleghi anche molto autorevoli avevano previsto che la malattia non sarebbe durata più di 2-3 mesi. Le dannose consegnenze di simili prognosi sul morale dei familiari, non meno che sulla reputazione del medico, sono di facile evidenza.
Un'ultima considerazione: bisogna evitare l'errore di trasferire a centri specialistici talora assai lontani, nell'illusione di impossibili guarigioni, leucemici gravissimi e già colpiti da ripetute recidive. Sono proprio questi i casi in cui l'apporto psicologico e umano del medico è indispensabile e risolutivo.
10. Conclusioni.
Da questo lungo discorso sulle leucemie si può trarre con certezza una conclusione: la prognosi delle leucemie acute, che fino a poco più di trent'anni fa erano mortali nel 100% dei casi, tanto che nessuna delle rarissime segnalazioni di guarigione di malati con asserita leucemia acuta aveva resistito alla critica, è ora notevolmente migliorata.
È ormai realtà indiscutibile che sono svariate centinaia i casi, registrati dalla letteratura più autorevole, di pazienti affetti da leucemia acuta guariti e tutto lascia sperare che a questi se ne aggiungeranno altri tra i pazienti che sono in completa remissione da oltre 5 anni.
La situazione migliorerà ulteriormente con il graduale progredire delle nostre conoscenze su molti problemi strettamente connessi con la terapia, alcuni dei quali, a prescindere dal più importante che è quello relativo all'eziologia ancora sconosciuta, possono così essere prospettati: 1) disporre di metodi per determinare la sensibilità delle cellule leucemiche ai vari agenti chemioterapici; 2) chiarire totalmente i meccanismi d'azione di molti dei farmaci impiegati; 3) riuscire a proteggere le cellule normali dai danni che su esse provocano i citostatici.
È comunque certo che attualmente nel campo della terapia delle leucemie non regna più il buio assoluto ma, com'è stato detto, s'incomincia a intravedere un barlume di luce all'estremo del tunnel.
Bibliografia.
A.I.L. (Italian Association against Leukaemia), A cooperative study on the therapy of acute lymphoblastic leukaemia, in ‟Haematologica", 1979, LXIV, pp. 119-147.
Aksoy, M., Dincol, K., Erdem, S., Dincol, G., Acute leukaemia due to chronic exposure to benzene, in ‟American journal of medicine", 1972, LII, pp. 160-166.
Alexander, P., Powles, R., Immunotherapy of human acute leukaemia, in ‟Clinics in haematology", 1978, VII, pp. 275-294.
Arlin, Z. A., Fried, J., Clarkson, B. D., Therapeutic role of cell kinetics in acute leukaemia, in ‟Clinics in haematology", 1978, VII, pp. 339-362.
Bahnson, C. B., Psychologic and emotional issues in cancer: the psychoterapeutic care of the cancer patient, in ‟Seminars in oncology", 1975, II, pp. 293-309.
Baikie, A. G., Chromosomes and leukaemia, in ‟Acta haematologica", 1966, XXXVI, pp. 157-173.
Baltimore, D., RNA-dependent DNA polymerase in virions of RNA tumor viruses, in ‟Nature", 1970, CCXXVI, pp. 1209-1211.
Baserga, A., Aspetti psicologici e familiari delle leucemie, in Le leucemie (a cura di A. Baserga), Roma 1972, pp. 963-976.
Bass, M., Clusters of leukaemia and Hodgkin's disease, in ‟New England journal of medicine", 1971, CCLXXXV, p. 461.
Battle, C., Bonfiglio, T. A., Miller, J. R., Pericarditis as the initial manifestation of acute leukaemia: report of a case, in ‟Journal of pediatrics", 1969, LXXV, pp. 692-694.
Belli, A., Belli, F., Il polmone nelle emolinfopatie maligne sistemiche. I. Studio introduttivo: inquadramento odierno della tematica e revisione critica della letteratura, in ‟Recenti progressi in medicina", 1980, LXIX, pp. 158-172.
Belli, F., Belli, A., Il polmone nelle emolinfopatie maligne sistemiche. II. Correlazione clinica, radiologica ed anatomopatologica nella storia evolutiva dei diversi quadri morbosi, in ‟Recenti progressi in medicina", 1980, LXIX, pp. 173-199.
Bennett, J. H., Two cases of disease and enlargement of the spleen in which death took place from the presence of purulent matter in the blood, in ‟Edinburgh medical and surgical journal", 1845, LXIV, pp. 413-423.
Bennett, J. M., Catovsky, D., Brennan, J., Morphologic and non-morphologic techniques in the characterization of leukaemic cells, in 18th Congress of the International Society of Hematology, Montreal 1980, pp. 28-32.
Bennett, J. M., Catovsky, D., Daniel, M. T., Flandrin, G., Galton, D. A. G., Gralnick, H. R., Sultan, C., Proposal for the classification of the acute leykaemias, in ‟British journal of haematology", 1976, XXXIII, pp. 451-458.
Bernard, J., Lévy, J.-P., Varet, B., Hématologie, Paris 1976.
Bessis, M., Brecher, G. (a cura di), Unclassificable leukemias, Berlin 1975.
Blackstock, A. M., Garson, O. M., Direct evidence for involvement of erythroid cells in acute myeloblastic leukaemia, in ‟Lancet", 1974, II, pp. 1178-1179.
Blatt, J., Reaman, G., Poplack, D. G., Biochemical markers in lymphoid malignancy, in ‟New England journal of medicine", 1980, CCCIII, pp. 918-922.
Bleufarb, S. M., Leukemia cutis, Springfield, Ill., 1960.
Bodey, G. P., Infections in hematological diseases, in ‟Clinics in haematology", 1976, V, 2, pp. 229-465.
Bodey, G. P., Rodriguez, V., Approaches to the treatment of acute leukemia and lymphoma in adults, in ‟Seminars in hematology", 1978, XV, pp. 221-261.
Bollum, F. J., Terminal deoxynucleotidyl transferase as a hematopoietic cell marker, in ‟Blood", 1979, LIV, pp. 1203-1215.
Bréton-Gorius, J., La leucemia megacarioblastica, in ‟Haematologica", 1979, LXIV, pp. 517-536.
Brody, R. S., Schottenfeld, D., Multiple primary cancers in Hodgkin's disease, in ‟Seminars in oncology", 1980, VII, pp. 187-201.
Brouet, J.-C., Preud'homme, J.-L., Seligman, M., The use of B and T membrane markers in the classification of human leukemias, with special reference to acute lymphoblastic leukemia, in ‟Blood cells", 1975, I, pp. 81-90.
Brouet, J.-C., Seligman, M., Chronic lymphocytic leukaemias as an immunoproliferative disorder, in ‟Clinics in haematology", 1977, VI, pp. 169-184.
Burch, P. R. J., Human cancer. Mendelian inheritance or vertical transmission?, in ‟Nature", 1963, CXCVII, pp. 1042-1045.
Canellos, G. P., The treatment of chronic granulocytic leukaemia, in ‟Clinics in haematology", 1977, VI, pp. 113-128.
Casciato, D. A., Scott, J. L., Acute leukemia following prolonged cytotoxic agent therapy, in ‟Medicine", 1979, LVIII, pp. 32-47.
Clarkson, B., Review of recent studies of cellular proliferation in acute leukemia, in ‟National Cancer Institute monograph", 1969, XXX, pp. 81-119.
Clarkson, B., Acute myelocytic leukemia in adults, in ‟Cancer", 1972, XXX, pp. 1572-1582.
Clarkson, B., Fried, J., Strife, A., Sakai, Y., Ota, K., Ohkita, T., Studies of cellular proliferation in human leukemia. III. Behaviour of leukemic cells in three adults with acute leukemia given continuous infusion of 3H-thymidine for 8 or 10 days, in ‟Cancer", 1970, XXV, pp. 1237-1260.
Cone, L., Uhr, J. W., Immunological deficiency disorders associated with chronic lymphocytic leukemia and multiple myeloma, in ‟Journal of clinical investigation", 1964, XLIII, pp. 2241-2248.
Cornes, J. R., Jones, T. G., Some important clinical and pathological features of leukaemic lesions in the gastro-intestinal tract, in ‟Proceedings of the Royal Society of Medicine", 1962, LV, pp. 702-703.
Coser, P., Quaglino, D., De Pasquale, A., Colombetti, V., Prinoth, O., Cytobiological and clinical aspects of tissue mast cell leukemia, in ‟British journal of haematology", 1980, XLV, pp. 5-12.
Court Brown, W. M., Doll, R., Mortality from cancer and other causes after radiotherapy for ankylosing spondylitis, in ‟British medical journal", 1965, II, pp. 1327-1332.
Creech, R. H., The psychologic support of the cancer patient: a medical oncologist's viewpoint, in ‟Seminars in oncology", 1975, II, pp. 285-292.
Dameshek, W., Chronic lymphocytic leukemia. An accumulative disease of immunologically incompetent lymphocytes, in ‟Blood", 1967, XXIX, pp. 566-584.
Di Guglielmo, G., Le eritremie, in Le emopatie (a cura di A. Ferrata), Milano 1923, pp. 112-167.
Di Guglielmo, G., Le malattie eritremiche ed eritroleucemiche, Roma 1962.
Dutcher, R. M., Viruses as etiological factors in leukemia. Comparative studies, in Perspectives in leukemia (a cura di W. Dameshek e R. M. Dutcher), New York 1968, pp. 13 ss.
Duttera, M. J., Bull, J. M., Northup, D., Henderson, E. S., Stashick, E. D., Carbone, P. P., Serial in vitro bone marrow culture in acute lymphocytic leukemia, in ‟Blood", 1973, XLII, pp. 687-699.
??? Editoriale: Leukemia and cytotoxic drugs, in ‟Lancet", 1971, I, pp. 70-71.
Egozine, J., Irwin, S., Maruffo, C. A., Chromosomal damage in LSD users, in ‟Journal of the American Medical Association", 1968, CCIV, pp. 214-218.
Ehrlich, P. (a cura di), Farbenanalytische Untersuchungen zur Histologie und Klinik des Blutes, Berlin 1891.
Evans, A. E., Gilbert, E. S., Zandstra, R., The increasing incidence of central nervous system leukemia in children (children's cancer study group A), in ‟Cancer", 1970, XXVI, pp. 404-414.
Faber, M., Johansen, C., Leukemia and other hematological diseases after thorotrast, in ‟Annals of the New York Academy of Sciences", 1967, CXLV, pp. 755-758.
Ferrata, A., Morfologia del sangue normale e patologico, Milano 1912.
Ferrata, A., Storti, E., Mauri, C., Malattie del sangue, Milano 19582.
Flandrin, G., Bernard, J., Cytological classification of acute leukemias. A survey of 1400 cases, in Unclassifiable leukemias (a cura di M. Bessis e G. Brecher), Berlin 1975, pp. 7-15.
Fliedner, T. M., Perry, S., Workshop on prognostic factors in human acute leukemia, in ‟Advances in the biosciences", 1975, XIV, pp. 3-203.
Fontana, G., Rizzo, S. C., Profilassi e terapia delle complicanze neuromeningee delle leucosi linfatiche acute, in Radiobiologia del linfocito (a cura di L. Oliva), Padova 1976, pp. 147-163.
Fontana, G., Rizzo, S. C., Ascari, E., Ricevuti, G., Balduini, C. L., Santagati, G., Profilassi e terapia delle complicanze neuromeningee delle leucosi linfatiche acute. Contributo casistico e revisione critica delle letteratura, in ‟Haematologica", 1975, LX, pp. 207-229.
Fontana, G., Rizzo, S. C., Balduini, C. L., Obesità diencefalica da localizzazione neuromeningea di leucosi linfatica acuta. Descrizione di un caso clinico, in ‟Haematologica", 1973, LVIII, pp. 1092-1098.
Fontana, G., Rizzo, S. C., Invernizzi, R., Ricevuti, G., Marini, G., L'impiego della citocentrifuga nella diagnostica delle complicanze neuromeningee delle leucosi acute, in ‟Rassegna clinico-scientifica", 1978, LIV, pp. 8-13.
Forni, A., Vigliani, E. C., Chemical leukemogenesis in man, in ‟Series haematologica", 1974, VII, pp. 211-223.
Fraumeni, J. F. Jr., Clinical epidemiology of leukemia, in ‟Seminars in hematology", 1969, VI, pp. 250-260.
Gale, R. P., Advances in the treatment of acute myelogenous leukemia, in ‟New England journal of medicine", 1979, CCC, pp. 1189-1199.
Gallo, R. C., Meyskens, F. L. Jr., Advances in the viral etiology of leukemia and lymphoma, in ‟Seminars in hematology", 1978, XV, pp. 379-398.
Gallo, R. C., Perry, S., Enzyme abnormality in human leukemia, in ‟Nature", 1968, CCXVIII, pp. 465-466.
Gallo, R. C., Yaung, S. S., Ting, R. C., RNA-dependent DNA polymerase of human acute leukaemic cells, in ‟Nature", 1970, CCXXVIII, pp. 927-929.
Galton, D. A. G., The pathogenesis of chronic lymphocytic leukemia, in ‟Canadian Medical Association journal", 1966, XCIV, pp. 1005-1010.
Galton, D. A. G., The chronic leukemias, in Blood and its disorders (a cura di R. M. Hardisty e D. J. Weatherall), Oxford 1974, pp. 952-992.
Galton, D. A. G., The chronic leukaemias, in Recent advances in hematology (a cura di A. V. Hoffbrand, M. C. Brain e J. Hirsh), Edinburgh 1977, pp. 219-242.
Galton, D. A. G., Dacie, J. V., Classification of the acute leukaemias, in Unclassificable leukemias (a cura di M. Bessis e G. Brecher), Berlin 1975, pp. 17-24.
Gavosto, F., Cell population growth in chronic myeloid leukaemia, in Proceedings of an International Symposium on chronic myeloid leukemia (a cura di S. Tura e M. Baccarani), Pavia 1972, pp. 67-75.
Gavosto, F., Kinetic conditions preventing the eradication of human leukemia, in ‟European journal of cancer", 1977, XIII, pp. 407-409.
Gavosto, F., Pileri, A., Gabutti, V., Masera, P., Cell population kinetics in human acute leukemia, in ‟European journal of cancer", 1967, III, pp. 301-307.
Gavosto, F., Pileri, A., Maraini, G., Proliferative capacity of acute leukaemia cells, in ‟Nature", 1960, CLXXXVII, pp. 611-612.
Geary, C. G., Catovsky, D., Wiltshow, E., Milner, G. R., Scholes, M. C., Van Noorden, S., Wadsworth, L. D., Muldal, S., MacIver, J. E., Galton, D. A. G., Chronic myelomonocytic leukaemia, in ‟British journal of hematology", 1975, XXX, pp. 289-301.
Ghione, M., Effetti tossici dei farmaci antitumorali sul sistema cardiovascolare, in ‟Recenti progressi in medicina", 1977, LXIII, pp. 382-410.
Gilliam, A. G., Age, sex and race selection at death from leukemia and lymphomas, in ‟Blood", 1953, VIII, pp. 693-702.
Golde, D. W., Cline, M. J., Regulation of granulopoiesis, in ‟New England journal of medicine", 1974, CCXCI, pp. 1388-1395.
Graham, D. C., Leukemia following X-ray therapy for ankylosing spondylitis, in ‟Archivs of internal medicine", 1960, CV, pp. 75-83.
Graham, S., Gibson, R., Lilienfeld, A., Shuman, L. M., Levin, M., Religion and ethnicity in leukemia, in ‟American journal of public health", 1970, LX, pp. 266-274.
Greeenberg, M. L., Chanana, A. D., Chronkite, E. P., Giacomelli, G., Rai, K. R., Schiffer, L. M., Stryckmans, P. A., Vincent, P. C., The generation time of human leukemic myeloblasts, in ‟Laboratory investigation", 1972, XXVI, pp. 245-252.
Gross, L., ‛Spontaneous' leukemia developing in C3H mice following inoculation in infancy with AK leukemic extracts of AK embryos, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1951, LXXVI, pp. 27-32.
Gross, L., Serial cell-free passage of radiation activated mouse leukaemia agent, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1959, C, pp. 102-105.
Gunz, F. W., Genetics of human leukaemia, in ‟Series haematologica", 1974, VII, pp. 164-191.
Gunz, F. W., Baikie, A. G. (a cura di), Leukemia, New York 19743.
Gunz, F. W., Vincent, P. C., Towards a cure of acute granulocytic leukemia?, in ‟Leukemia research", 1977, I, pp. 51-66.
Hansen, M. M., Chronic lymphocytic leukemia: clinical studies based on 189 cases followed for a long time, in ‟Scandinavian journal of haematology", 1978, suppl. 18, pp. 2-78.
Hardisty, R. M., Weatherall, D. J. (a cura di), Blood and its disorders, Oxford 1974.
Harousseau, J. L., Tabelem, G., Schaison, G., Chastang, C., Auclerc, M. F., Weil, M., Jacquillat, C., Bernard, J., High risk acute lymphocytic leukemia. A study of 141 cases with high blasts counts, in 17th Congress of the International Society of Haematology. Abstracts, vol. I, Paris 1979, p. 252.
Hayhoe, F. G. J., Quaglino, D., Haematological cytochemistry, Edinburgh 1980.
Henderson, E. S., Wallace, J. Y., Scharlow, C., Rakowski, I., Ellison, R. R., Holland, J. F., Factors influencing prognosis in adult acute myelocytic leukemia, in ‟Advances in the biosciences", 1975, XIV, pp. 71-82.
Hillestad, L. K., Acute promyelocytic leukemia, in ‟Acta medica scandinava", 1957, CLIX, pp. 189-194.
Hirschhorn, K., Cytogenetic alterations in leukemia, in Perspectives in leukemia (a cura di W. Dameshek e R. M. Dutcher), New York 1968, pp. 113 ss.
Holland, J. F., Epidemic acute leukemia, in ‟New England journal of medicine", 1970, CCLXXXIII, pp. 1165-1166.
Hustu, H. O., Aur, R. J. A., Extramedullary leukaemia, in ‟Clinics in haematology", 1978, VII, pp. 313-337.
Hyman, C. B., Bogle, J. M., Brubaker, C. A., Williams, K., Hammond, D., Central nervous system involvement by leukemia in children. I. Relationship to systemic leukemia and description of clinical and laboratory manifestations, in ‟Blood", 1965, XXV, pp. 1-12.
Ichimaru, M., Ishimaru, T., Belsky, J. L., Incidence of leukemia in atomic bomb survivors belonging to a fixed cohort in Hiroshima and Nagasaki, 1950-71. Radiation dose, years after exposure, age at exposure, and type of leukemia, in ‟Japanese journal of radiation research", 1978, XIX, pp. 262-276.
Ihle, J. N., Experimental models and conceptual approaches to studies of lymphomas and leukemia: etiology, biology and control, in ‟Seminars in hematology", 1978, XV, pp. 95-115.
Invernizzi, R., Marini, G., Ippoliti, G., Casirola, G., Ascari, E., Comparison between cytological and cytochemical classification of acute leukaemia, in Therapy of acute leukemias (a cura di F. Mandelli), Roma 1979, pp. 60-65.
Ishimaru, T., Otake, M., Ichimaru, M., Dose-response relationship of neutrons and γ rays to leukemia incidence among atomic bomb survivors in Hiroshima and Nagsaki by type of leukemia, 1950-1971, in ‟Radiation research", 1979, LXXVII, pp. 377-394.
Jaffe, N., Metabolic and biochemical changes in leukemia, in Acute childhood leukemia (a cura di C. Pochedley), Basel 1975, pp. 113-136.
Jaffe, N., Traggis, D. F., Tefet, M., Acute leukemia presenting with pericardial tamponade, in ‟Pediatrics", 1970, XLV, pp. 461-465.
Jarrett, W. F. H., Crowford, E. M., Martini, W. B., Davie, F., Leukaemia in the cat. A virus-like particle associated with leukaemia (lymphosarcoma), in ‟Nature", 1964, CCII, pp. 557-568.
Jensen, M. K., Killmann, S.-A., Chromosome studies in acute leukaemia: evidence for chromosomal abnormalities common to erythroblasts and leukaemic white cells, in ‟Acta medica scandinava", 1967, CLXXXI, pp. 47-53.
Johnson, R. E., Radiotherapy as primary treatment for chronic lymphocytic leukaemia, in ‟Clinics in haematology", 1977, VI, pp. 237-244.
Killmann, S.-A., Chronic myelogenous leukemia: preleukemia or leukemia?, in Proceedings of an International Symposium on chronic myeloid leukaemia (a cura di S. Tura e M. Baccarani), Pavia 1972, pp. 45-53.
Kirshbaum, J. D., Preuss, F. S., Leukemia: a clinical and pathologic study of 123 fatal cases in a series of 14.400 necropsies, in ‟Archives of internal medicine", 1943, LXXI, pp. 777-792.
Krebs, C., Wagner, A., Rask-Neilson, H. C., The origin of lymphosarcomatosis and its relations to other forms of leucosis in white mice, in ‟Acta radiologica", 1930, suppl. 10, pp. 2-54.
Land, C. E., Estimating cancer risks from low doses of ionizing radiation, in ‟Science", 1980, CCIX, pp. 1197-1203.
Laszlo, J., Rundles, R. W., Morphology of granulocytes, in Hematology (a cura di W. J. Williams, E. Beutler, A. J. Erslev e R. W. Rundles), New York 19772, pp. 659 ss.
Lee, R. E., Ellis, L. D., The storage cells of chronic myelogenous leukemia, in ‟Laboratory investigation", 1971, XXIV, pp. 261-264.
Leef, F., Kende, G., Ramot, B., Testicular leukemia, in ‟Pediatrics", 1970, XLV, pp. 338-339.
Levine, A. S., Schimpff, S. C., Graw, R. G. Jr., Young, R. C., Hematologic malignancies and other marrow failure states: progress in the management of complicating infections, in ‟Seminars in hematology", 1974, XI, pp. 141-202.
Lissauer, H., Zwei Fälle von Leukämie, in ‟Berliner klinische Wochenschrift", 1865, II, pp. 403-404.
Lister, T. A., Yankee, R. A., Blood component therapy, in ‟Clinics in haematology", 1978, VII, pp. 407-423.
Lyon, J. L., Klauber, M. R., Gardner, J. W., Udall, K. S., Childhood leukemias associated with follout from nuclear testing, in ‟New England journal of medicine", 1979, CCC, pp. 397-402.
McAndrew, G. M., Ogston, D., Dawson, A. A., An evaluation of sternal tenderness as a sign of leukaemia, in ‟Acta haematologica", 1970, XLIII, pp. 309-312.
MacMahon, B., Koller, E. K., Ethnic differences in the incidence of leukemia, in ‟Blood", 1957, XII, pp. 1-10.
Mason, T. E., Demaree, R. S., Margolis, C. I., Granulocytic sarcoma (chloroma), two years preceding myelogenous leukemia, in ‟Cancer", 1973, XXXI, pp. 423-432.
Mauer, A. M., Cell kinetics and practical consequences for therapy of acute leukemia, in ‟New England journal of medicine", 1975, CCXCIII, pp. 389-393.
Mauer, A. M., Therapy of acute lymphoblastic leukemia in childhood, in ‟Blood", 1980, LVI, pp. 1-10.
Mauer, A. M., Fisher, V., Comparison of the proliferative capacity of acute leukaemia cells in bone marrow and blood, in ‟Nature", 1962, CXCIII, pp. 1085-1086.
Maugeri, S., Pollini, G., Le leucemie da tossici industriali, in Le leucemie (a cura di A. Baserga), Roma 1972, pp. 153-171.
Mauri, C., Localizzazioni toraciche delle emolinfopatie maligne, in Le malattie dell'apparato respiratorio (a cura di G. Spina e G. Bonsignore), vol. II, Torino 1978, pp. 487-511.
Mauri, C., Malattie del sangue e degli organi ematopoietici, in Trattato di patologia medica (a cura di U. Teodori), Roma 19803, p. 1601.
Mauri, C., Torelli, U., Di Prisco, U., Silingardi, V., Artusi, T., Emilia, G., Lymphoid blastic crisis at the onset of chronic granulocytic leukemia. Report of two cases, in ‟Cancer", 1977, XL, pp. 865-870.
Mavligit, G. M., Stuckey, S. E., Cabanillas, F. F., Keating, M. J., Tourtellotte, W. W., Schold, S. C., Freireich, E. J., Diagnosis of leukemia or lymphoma in the central nervous system by betamicroglobulin determination, in ‟New England journal of medicine", 1980, CCCIII, pp. 718-722.
Mertelsman, R. Thaler, H. T., To, L., Gee, T. S., McKenzie, S., Schauer, P., Friedman, A., Arlin, Z., Cirrincione, C., Clarkson, B., Morphological classification, response to therapy and survival in 263 adult patients with acute nonlymphoblastic leukemias, in ‟Blood", 1980, LVI, pp. 773-781.
Metzgar, R. S., Mohanakumar, T., Tumor-associated antigens of human leukemia cells, in ‟Seminars in hematology", 1978, XV, pp. 139-156.
Milham, S., Leukemia in husbands and wives, in ‟Science", 1965, CXLVIII, pp. 98-100.
Miller, D. G., Pattern of immunological deficiency in lymphomas and leukemias, in ‟Annals of internal medicine", 1962, LVII, pp. 703-716.
Miller, R. W., Down's syndrome (mongolism), other congenital malformations and cancers among the sibs of leukemic children, in ‟New England journal of medicine", 1963, CCLXVIII, pp. 393-401.
Miller, R. W., Relation between cancer and congenital defects, an epidemiologic evaluation, in ‟Journal of the National Cancer Institute", 1968, XL, pp. 1079-1085.
Minot, G. R., Buckman, T. E., Isaacs, R., Chronic myelogenous leukemia, in ‟Journal of the American Medical Association", 1924, LXXXII, pp. 1489-1494.
Montuori, R., La meningosi leucemica, Pisa 1977.
Moore, M. A. S., Marrow culture. A new approach to the classification of leukemias, in ‟Blood cells", 1975, I, pp. 149-158.
Moore, M. A. S., Spitze, G., Williams, N., Metcalf, D., Buckley, J., Agar culture studies in 127 cases of untreated acute leukemias: the prognostic value of reclassification of leukemia according to in vitro growth characteristics, in ‟Blood", 1974, XLIV, pp. 1-18.
Mullin, B. R., Harris, J. W., Chloroma (granulocytic sarcoma), in ‟American journal of the medical sciences", 1972, CCLXIV, pp. 415-422.
Neumann, E., Über myelogene Leukämie, in ‟Berliner klinische Wochenschrift", 1878, XV, pp. 69-72.
Pear, B. L., Skeletal manifestations of the lymphomas and leukemias, in ‟Seminars in roentgenology", 1974, IX, pp. 229-240.
Phillips, E. A., Kempin, S., Passe, S., Mike, V., Clarkson, B., Prognostic factors in chronic lymphocytic leukaemia and their implications for therapy, in ‟Clinics in haematology", 1977, VI, pp. 203-222.
Pochedley, C., Leukemia and lymphoma in the nervous system, Springfield, Ill., 1977.
Polli, E., La leucemia, biologia e clinica, Padova 1967.
Priest, J. R., Ramsay, N. K., Latchaw, R. E., Lockman, L. A., Hosegawa, D. K., Coates, T. D., Coccia, P. F., Edson, J. R., Nesbit, M. E., Krivit, W., Thrombotic and hemorrhagic strokes complicating early therapy for childhood acute lymphoblastic leukemia, in ‟Cancer", 1980, XLVI, pp. 1548-1554.
Pusey, W. A., Report of cases treated with Roentgen rays, in ‟Journal of the American Medical Association", 1902, XXXVIII, pp. 911-919.
Rai, K. R., Sawitsky, A., Cronkite, E. P., Channa, A. D., Levy, R. N., Pasternack, B. S., Clinical staging of chronic lymphocitic leukemia, in ‟Blood", 1975, XLVI, pp. 219-234.
Ravich, R. B., Reed, C. S., Stephens, F. O., Vincent, P. C., Gunz, F. W., Spontaneous rupture of the spleen in acute myeloid leukemia, in ‟The medical journal of Australia", 1971, I, pp. 90-92.
Reardon, G., Moloney, W. C., Chloroma and related myeloblastic neoplasms, in ‟Archives of internal medicine", 1961, CVIII, pp. 864-871.
Rees, J. K. H., Sandler, R. M., Challener, J., Hayhoe, F. G. J., Treatment of acute myeloid leukaemia with a triple cytotoxic regime: DAT, in ‟British journal of cancer", 1977, XXXVI, pp. 770-776.
Rizzo, S. C., Balduini, C. L., Beviglia, E., Gobbi, P. G., Ricevuti, G., Fontana, G., Chronic granulocytic leukemia. A retrospective study of 91 cases. I. Hemogram at diagnosis and research of prognostic factors, in ‟Haematologica", 1979, LXIV, pp. 777-785.
Rizzo, S. C., Balduini, C. L., Invernizzi, R., Ricevuti, G., Fontana, G., Ascari, E., Prognostic factors and treatment response in acute lymphoblastic leukemia of children and adults treated with a non aggressive combination chemotherapy, in ‟Haematologica", 1980, LXV, pp. 70-81.
Rizzo, S. C., Fontana, G., Balduini, C. L., Ricevuti, G., Osteolytic lesions in chronic granulocytic leukemia: a report of four cases, in ‟Heamatologica", 1977, LXII, pp. 61-74.
Rizzo, S. C., Fontana, G., Marini, G., Balduini, C. L., Marked eosinophilia associated with acute lymphoblastic leukemia, in ‟Haematologica", 1976, LXI, pp. 81-85.
Roath, S., Observations on the etiology of acute leukaemia, in ‟Clinics in haematology", 1972, I, pp. 23-47.
Roath, S., Israels, M. C. G., Wilkinson, J. F., The acute leukaemias: a study of 580 patients, in ‟Quarterly journal of medicine", 1964, XXXIII, pp. 257-283.
Roberts, W. C., Bodey, G. P., Wertlake, P. I., The heart in acute leukemia: a study of 420 autopsy cases, in ‟American journal of cardiology", 1968, XXI, pp. 388-412.
Rosner, F., Grünwald, H. S., Cytotoxic drugs and leukaemogenesis, in ‟Clinics in haematology", 1980, IX, pp. 663-681.
Rossi, A., Sebastiani, A., Il quadro oculare delle leucemie, in Le leucemie (a cura di A. Baserga), Roma 1972, pp. 581-594.
Rowley, J. D., Chromosomes in leukemia and lymphoma, in ‟Seminars in hematology", 1978, XV, pp. 301-319.
Rowley, J. D., The cytogenetics of acute leukaemia, in ‟Clinics in haematology", 1978, VII, pp. 385-406.
Rowley, J. D., Ph1-positive leukaemia, including chronic myelogenous leukaemia, in ‟Clinics in haematology", 1980, IX, pp. 55-86.
Sandberg, A. A., Cytogenetics of hematological disorders, in 18th Congress of the International Society of Hematology, Montreal 1980, pp. 32-36.
Sanders, J. E., Thomas, E. D., Bone marrow transplantation for acute leukaemia, in ‟Clinics in haematology", 1978, VII, pp. 295-311.
Saunders, E. F., Lampkin, B. C., Mauer, A. M., Variation of proliferative activity in leukemic cell populations of patients with acute leukemia, in ‟Journal of clinical investigation", 1967, XLVI, pp. 1356-1363.
Saunders, E. F., Mauer, A. M., Reentry of nondividing leukemic cells into a proliferative phase in acute childhood leukemia, in ‟Journal of clinical investigation", 1969, XLVIII, pp. 1299-1305.
Schaison, G., Jaquillat, C., Tanzer, J., Weil, M., Gemon, M. F., Les méningites leucémiques, in ‟La revue du praticien", 1973, XXIII, pp. 185-193.
Segi, M., Kurihara, M., Cancer mortality for selected sites in 24 countries (1962-1963), Sendai 1966.
Shanbrom, E., Finch, S. C., The auditory manifestations of leukemia, in ‟Yale journal of biology and medicine", 1958, XXXI, pp. 144-156.
Shifrine, M., Bulgin, M. S., Dollaahide, N. Z., Wolf, H. C., Taylor, N. J., Wilson, F. D., Transplantation of radiation-induced canine myelomonocytic leukaemia, in ‟Nature", 1971, CCXXXII, pp. 405-406.
Simone, J. V., Acute lymphocytic leukemia in childhood, in ‟Seminars in hematology", 1974, XI, pp. 25-39.
Simone, J. V., The treatment of acute lymphoblastic leukaemia, in ‟British journal of haematology", 1980, XLV, pp. 1-4.
Skipper, H. E., Schabel, F. M. Jr., Lloyd, H., Experimental therapeutics and kinetics: selection and overgrowth of specifically and permanently drug-resistant tumor cell, in ‟Seminars in hematology", 1978, XV, pp. 207-219.
Sokal, G., Michaux, J. L., Van den Berghe, H., The karyotype in refractory anaemia and pre-leukaemia, in ‟Clinics in haematology", 1980, IX, pp. 129-139.
Sokal, J. E., Immunotherapy for chronic granulocytic leukaemia, in ‟Clinics in haematology", 1977, VI, pp. 129-139.
Spiegelman, S., Molecular evidence for the association of RNA tumor viruses with human mesenchymal malignancies, in Modern trends in human leukemia (a cura di R. Neth e altri), vol. II, München 1976, pp. 391 ss.
Spiers, A. S. D., Metamorphosis of chronic granulocytic leukemia: diagnosis, classification and management, in ‟British journal of haematology", 1979, XLI, pp. 1-7.
Spilling, E., Über Blutuntersuchungen bei Leukämie, in Farbenanalytische Untersuchungen zur Histologie und Klinik des Blutes (a cura di P. Ehrlich), Berlin 1891, pp. 54-78.
Spitze, G., Verna, D. S., Dicke, K. A., McCredie, K. B., Culture studies in vitro in human leukemia, in ‟Seminars in hematology", 1978, XV, pp. 352-378.
Sternby, N. H., Studies in enlargement of leukaemic kidneys, in ‟Acta haematologica", 1955, XIV, pp. 354-362.
Storti, E., The history of leukaemia therapy, in ‟Haematologica", 1978, LXIII, pp. 74-86.
Storti, E., Leucemia a cellule capellute o leucemia a hairy-cell o tricoleucemia, in ‟Haematologica", 1981, LXVI, pp. 835-878.
Straus, D. J., Mertelsmann, R., Koziner, B., McKenzie, S., De Harven, E., Arlin, Z. A., Kempin, S., Broxmeyer, H., Moore, M. A. S., Menendez-Botet, C. J., Gee, T. S., Clarkson, B. D., The acute monocytic leukemias: multidisciplinary studies in 45 patients, in ‟Medicine", 1980, LIX, pp. 409-425.
Stryckmans, P. A., Debusscher, L., Collard, E., Cell kinetics in chronic granulocytic leukaemia, in ‟Clinics in haematology", 1977, VI, pp. 21-40.
Stryckmans, P.A., Debusscher, L., Collard, E., Cell kinetics in chronic lymphocytic leukaemia, in ‟Clinics in haematology", 1977, VI, pp. 159-167.
Sultan, C., Dysmyelopoietic syndrome, in Classification of acute leukemia (a cura di H. R. Gralnick), in ‟Annals of internal medicine", 1977, LXXXVII, pp. 740-753.
Temin, H.M., Mizutani, S., RNA-dependent DNA polymerases in virions of Rous sarcoma virus, in ‟Nature", 1970, CCXXVI, pp. 1211-1213.
Thomas, L. B., Forkner, C. E., Frei, E. III, Besse, B. E., Stabeau, J. R., The skeletal lesions of acute leukemia, in ‟Cancer", 1961, XIV, pp. 608-621.
Thompson, R. B., Walker, W., Study of 50 cases of acute leukaemia in childhood, in ‟British medical journal", 1962, I, pp. 1165-1169.
Tjalma, R. A., Implications of animal cancers to human neoplasia. Epidemiologic considerations, in ‟International journal of cancer", 1968, III, pp. 1-6.
Todaro, G., Evidence for the interspecies transmission of tumor virus genes, in Recent advances in cancer research: cell biology, molecular virology and tumor virology (a cura di R. C. Gallo), Cleveland 1977, pp. 51 ss.
Tomonaga, M., Leukaemia in Nagasaki atomic bomb survivors from 1945 through 1959, in ‟Bulletin of the World Health Organisation", 1962, XXVI, pp. 619-631.
Velpeau, A., Sur la résorption du pus et sur l'altération du sang dans les maladies, in ‟Revue médicale", 1827, II, pp. 216-240.
Videbaek, A., Heredity in human leukemia and its relation to cancer, København 1947.
Vincent, P. C., Cell kinetics of the leukemias, in Leukemia (a cura di F. W. Gunz e A. G. Baikie), New York 19743, pp. 189-221.
Virchow, R., Weisses Blut, in ‟Froriep Notizen", 1845, XXXVI, pp. 151-156.
Virchow, R., Weisses Blut und Milztumoren, in ‟Medizinische Zeitung", 1847, XVI, pp. 9-15.
Virchow, R., Die Leukämie, in Gesammelte Abhandlungen zur wissenschaftlichen Medizin, Frankfurt a. M. 1856, pp. 190-211.
Wachstein, M., Alkaline phosphatase activity in normal and abnormal human blood and bone marrow, in ‟Journal of laboratory and clinical medicine", 1946, XXXI, pp. 1-17.
Warren, S., Radiation carcinogenesis, in ‟Bulletin of the New York Academy of Medicine", 1970, XLVI, pp. 131-147.
Watanabe, S., On the incidence of leukemias in Hiroshima during the past fifteen years from 1946 to 1960, in Proceedings of the 9th Congress of the International Society of Hematology, vol. I, Ciudad de México 1964, p. 511.
Weiss, H. D., Walker, M. D., Wiernik, P. H., Neurotoxicity of commonly used antineoplastic agents, in ‟New England journal of medicine", 1974, CCXCI, pp. 75-81 e 127-133.
Whittaker, J. A., Advances in the management of adulte acute myelogenous leukaemia, in ‟British medical journal", 1980, II, pp. 960-964.
Whittaker, J. A., Immunotherapy in the treatment of acute leukaemia, in ‟British journal of haematology", 1980, XLV, pp. 187-193.
Whittaker, J. A., Withey, J., Powell, D. E. B., Parry, T. E., Khurshid, M., Leukaemia classification: a study of the accuracy of diagnosis in 456 patients, in ‟British journal of hematology", 1979, XLI, pp. 177-184.
Wiernik, P. H., Treatment of acute leukaemia in adult, in ‟Clinics in haematology", 1978, VII, pp. 259-273.
Williams, W. J., Beutler, E., Erslev, A. J., Rundles, R. W. (a cura di), Hematology, New York 19772.
Wiltshow, E., Chemotherapy in chronic lymphocytic leukaemia, in ‟Clinics in haematology", 1977, VI, pp. 223-235.
Wintrobe, M. M., Clinical haematology, Philadelphia 19747.
Woodruff, R. K., Malpas, J. S., Paxton, A. M., Lister, T. A., Plasma cell leukemia (PCL): a report of 15 patients, in ‟Blood", 1978, LII, pp. 839-845.
Yodoi, J., Takatsuki, K., Masuda, T., Two cases of T cell chronic lymphocytic leukemia in Japan, in ‟New England journal of medicine", 1974, CCXC, pp. 572-573.
Zuelzer, W. W., Cox, D. E., Genetic aspects of leukemia, in ‟Seminars in hematology", 1969, VI, pp. 228-249.