
Oncogeni
Oncogeni
(App. V, iii, p. 763)
La ricerca oncologica negli anni Novanta si è focalizzata sull'identificazione e l'isolamento dei geni responsabili delle sindromi cancerose ereditarie. Anche se la maggioranza delle mutazioni responsabili di tumori è di origine somatica, cioè solo il DNA delle cellule cancerose dell'individuo porta la mutazione nel gene responsabile, circa l'1% dei tumori ha una trasmissione ereditaria. In questi casi, il DNA di tutte le cellule di quel particolare individuo porta la mutazione nel gene d'interesse. La scoperta dei geni dei tumori ereditari ha rappresentato ed è ancora oggi una grande sfida per la ricerca scientifica, non solo perché ha consentito di mettere a punto nuovi approcci e strategie sperimentali, ma anche perché ha permesso di chiarire ulteriormente il ruolo che gli o. svolgono nel normale funzionamento della cellula, nei meccanismi di trasduzione di segnali extracellulari e di come mutazioni siano in grado di alterarli.
Infatti, una volta che l'anamnesi familiare consente di affermare che un determinato soggetto è presumibilmente affetto da tumore ereditario, si esegue un'analisi di linkage genetico: lo studio di diverse generazioni di grandi famiglie consente di stabilire che alcuni marcatori genetici (markers), localizzati su una particolare regione cromosomica, si trasmettono alla progenie assieme al presunto gene responsabile del cancro (cosegregazione). In questo modo, il gene di interesse viene localizzato su una regione di un determinato cromosoma, e viene più precisamente mappato per successive associazioni con markers polimorfici. Questi ultimi risiedono in geni noti o corrispondono a DNA 'anonimo' localizzato nella stessa regione. Il gene viene poi identificato e isolato con la tecnica del clonaggio di posizione (positional cloning) o con la tecnica più recente delle EST (Expressed Sequence Tag), sequenze espresse e facilmente amplificabili tramite PCR (Polymerase Chain Reaction). Le EST sono sequenze conservate presso banche dati che si ottengono dall'analisi e dal sequenziamento sistematico e casuale del maggior numero di cloni di cDNA derivati per copia dalle popolazioni di geni espressi (mRNA; v. biologia molecolare, App. V e in questa Appendice). Tali cloni derivano da librerie di tessuti particolari o di fasi diverse dello sviluppo embrionale o da regioni specifiche di cromosomi. Quest'analisi consente di ottenere ulteriori informazioni sulla specificità tissutale di espressione del nostro clone o sulla sua espressione nel corso dello sviluppo. La tappa finale di questa indagine è costituita dalla ricerca della mutazione, presente solo nel gene dei soggetti affetti. Infine, identificato l'allele mutato, è possibile confermare la sua attività trasformante mediante saggi funzionali in vitro.
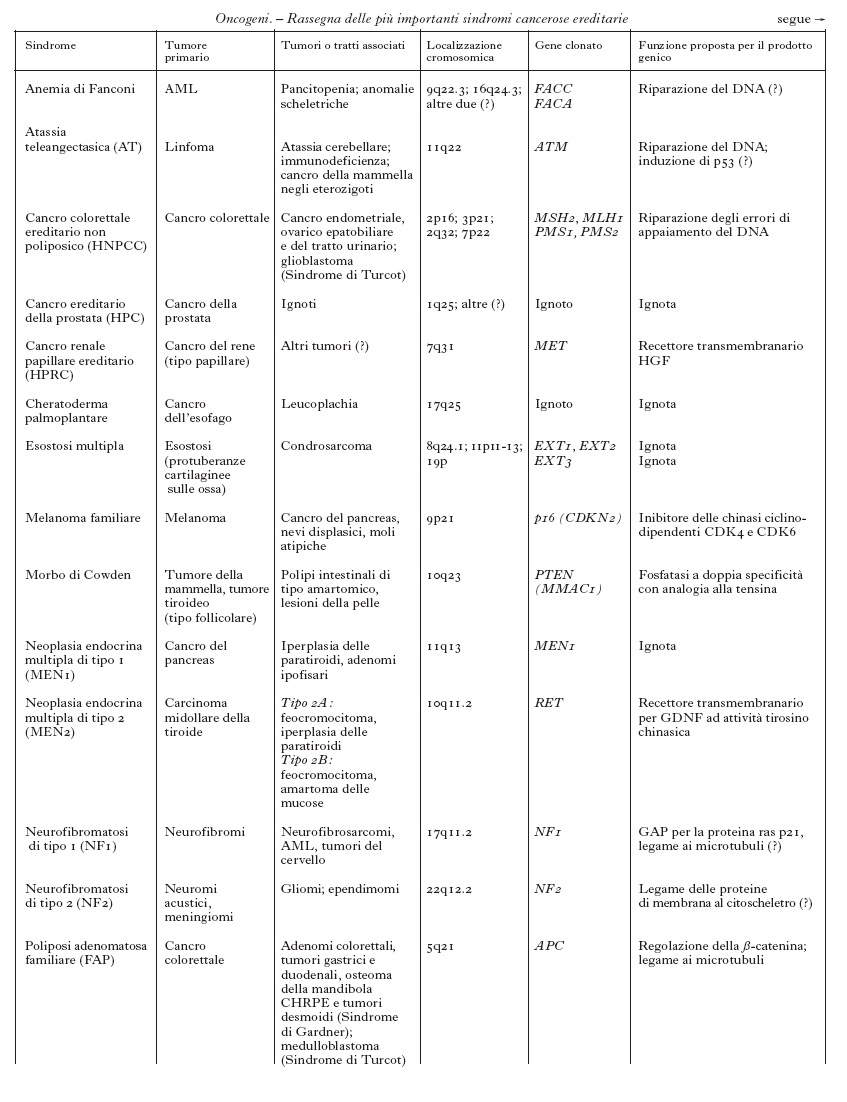
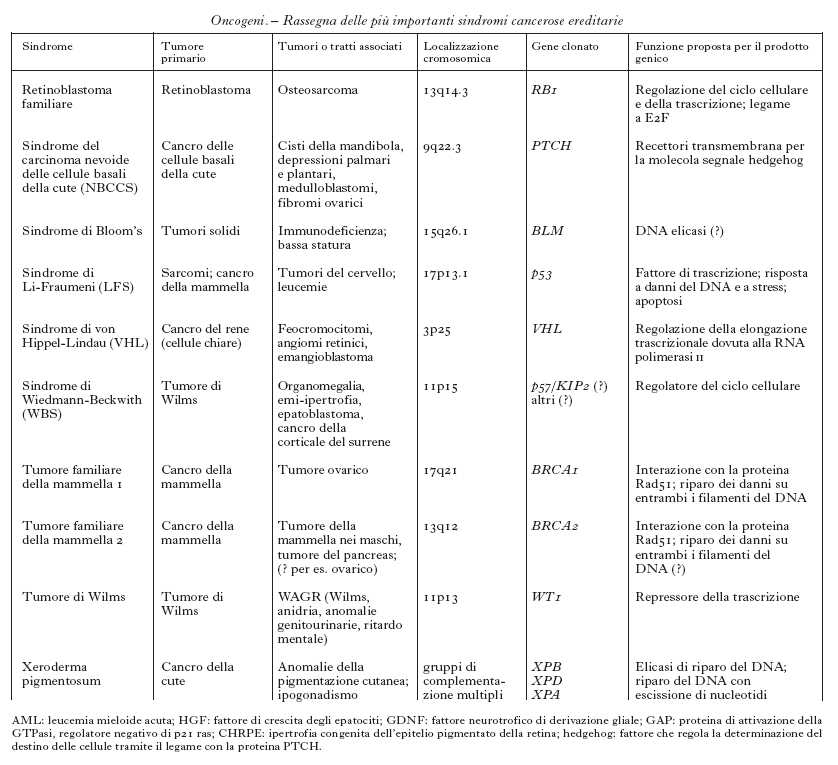
Anche se il numero complessivo dei tumori trasmessi ereditariamente è ancora sconosciuto, sono state definite almeno 20 forme diverse di tumori ereditari e ne sono stati identificati i geni responsabili (v. tabb. 1a e 1b). La ricerca di queste forme ereditarie è resa più difficile dal fenomeno della penetranza incompleta dei geni, cioè dalla diversa probabilità con cui un individuo portatore di un gene mutato può sviluppare un tumore: questa diversa probabilità dipende dalla particolare mutazione del gene responsabile. Ulteriori difficoltà si verificano per quegli individui che, pur presentando il quadro tipico della malattia, non portano la mutazione nel gene (fenocopie). Queste incertezze diagnostiche hanno messo in evidenza che il quadro clinico con cui si manifesta un tumore ereditario è la risultante di diversi fattori patogenetici, quali stile di vita, alimentazione e qualità dell'ambiente, in grado di modificarne l'andamento. Un ruolo importante è svolto da quei geni definiti modificatori, che una volta mutati possono alterare la progressione del tumore. A seconda del tipo, del numero e dell'ordine con cui partecipano all'instaurarsi o al progredire del processo neoplastico, corrisponde un andamento più o meno aggressivo del tumore. Alcuni di questi geni sono noti singolarmente sia come o. sia come geni soppressori di tumore; altri determinano instabilità del DNA cosiddetto satellite e/o sono essi stessi responsabili dell'integrità del DNA cromosomico. Un esempio di gene modificatore è il gene della telomerasi, l'enzima responsabile del mantenimento dell'integrità delle estremità dei cromosomi: la sua inattivazione è stata associata alla progressione tumorale perché renderebbe i cromosomi più instabili e facilmente degradabili.
La maggioranza dei geni modificatori è ancora del tutto sconosciuta. Un importante obiettivo della ricerca nei primi anni del Duemila sarà identificarli e determinarne il coinvolgimento nelle varie forme tumorali. Un esempio particolarmente interessante del ruolo che potrebbero svolgere tali geni è quello rappresentato dai tumori causati dall'o. RET. È questo un gene cellulare (proto-o.) che funziona come recettore di membrana con attività tirosino-chinasica sulla superficie delle cellule derivate dalla cresta neurale: interagisce con uno specifico ligando, il fattore neurotrofico derivato dalle cellule gliali (GDNF, Glial Derived Nerve Factor), innescando una serie di eventi che portano alla divisione della cellula bersaglio. In seguito a mutazioni puntiformi, il proto-o. RET si trasforma in o. dominante, cioè in grado di formare tumori e, infatti, forme mutate sono responsabili di alcune sindromi tumorali ereditarie trasmesse con meccanismo autosomico dominante: la sindrome endocrina multipla (MEN, Multiple Endocrine Neoplasia) di tipo 2A caratterizzata da tumore midollare della tiroide, tumore della midollare del surrene (feocromocitoma) e iperplasia delle paratiroidi; la sindrome MEN 2B in cui al tumore midollare della tiroide e al feocromocitoma si associano ganglioneuromi del tratto gastroenterico e alterazioni morfologiche di tipo marfanoide senza interessamento delle paratiroidi. Mutazioni puntiformi del proto-o. RET sono responsabili di un'altra forma tumorale in cui il tumore midollare della tiroide è l'unica manifestazione clinica della malattia, il cosiddetto tumore midollare della tiroide familiare (FMTC, Familiar Medullar Tyroid Carcinoma). Poiché mutazioni degli stessi codoni del proto-o. RET sono responsabili sia di casi di MEN 2A sia di molti FMTC, riesce difficile associare alle stesse mutazioni quadri clinici così diversi per insorgenza, decorso e prognosi. Inoltre, mutazioni di RET, localizzate in regioni diverse del gene, sono responsabili della MEN 2A e della MEN 2B, sindromi molto diverse per insorgenza e andamento clinico. Queste considerazioni fanno ritenere che le mutazioni del proto-o. RET non sono da sole sufficienti a determinare l'espressione completa delle sindromi tumorali a esse associate. È verosimile che alle mutazioni di RET, evento scatenante della proliferazione cellulare, si debbano aggiungere mutazioni in altri geni denominati modificatori, secondo il concetto che il processo tumorale progredisce a tappe. Il quadro clinico finale è, pertanto, la risultante dell'intervento di questi vari cofattori. Ancora più interessante è l'osservazione che alcune mutazioni legate a MEN 2A e FMTC si ritrovino anche in casi di megacolon congenito o aganglionosi del colon, una malattia dei neonati e dei bambini caratterizzata da alterata peristalsi di un tratto del colon con conseguente espansione dell'intestino a monte. Questa malattia non è una forma tumorale e si ritiene che sia dovuta a perdita di funzione di RET. In questi casi si realizza una situazione in cui mutazioni di tipo attivante, responsabili di quadri clinici da guadagno di funzione (tumori), si ritrovano associate a un quadro clinico da perdita di funzione. Per conciliare i dati clinici con i dati genetici è verosimile ritenere che geni modificatori differenti possano intervenire a vari tempi e in vario numero nella progressione del processo neoplastico, interessando organi diversi e determinando i diversi quadri clinici descritti. Questo tipo di argomentazione è confermato in parte dal fatto che una certa percentuale di tumori midollari della tiroide sporadici sono causati dalla stessa mutazione responsabile della MEN 2B.
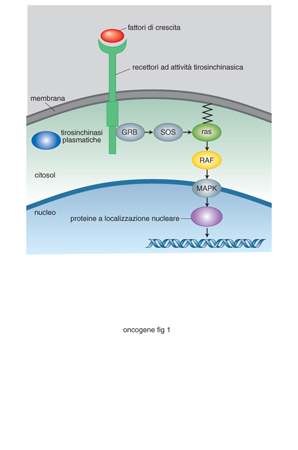
Un'altra importante osservazione, scaturita da questa ricerca sui geni responsabili di tumori ereditari, è che una certa percentuale di tumori sporadici sono provocati da mutazioni negli stessi geni responsabili delle forme familiari. È possibile quindi ritenere che, almeno in alcuni casi, meccanismi patogenetici simili siano responsabili sia di tumori sporadici che di tumori familiari. Questa similarità ha rappresentato un ulteriore stimolo a delucidare i meccanismi con cui singoli proto-o. funzionano nel metabolismo cellulare, considerando che solo le forme attivate hanno attività tumorigenica. A tutt'oggi, sono stati identificati circa 100 o. che possono essere classificati in diversi gruppi, rappresentativi di diversi tipi di attività (fig. 1): da proteine chinasi di membrana a proteine chinasi non recettoriali, da fattori del ciclo cellulare a fattori trascrizionali, da regolatori della trascrizione a proteine del riparo del DNA. Da questa classificazione è possibile prevedere il numero delle funzioni modificate e l'entità degli effetti di una loro mutazione a seconda della posizione che i singoli proto-o. occupano lungo le varie vie di trasduzione del segnale. Se la via di trasduzione è lineare, tale per cui un segnale passa direttamente da un componente al successivo, il risultato finale è lo stesso, indipendentemente dalla posizione che il componente mutato occupa lungo la via stessa: una mutazione costitutiva, cioè, fa sì che quel fattore non necessiti di essere attivato dal segnale proveniente da un componente più a monte. Molto più frequentemente, le vie di trasduzione si ramificano a più livelli, in modo che il segnale iniziale attivi una serie di vie secondarie e produca una molteplicità di risposte differenti (pleiotropicità). In questi casi, mutazioni dei componenti più a valle sono responsabili di un numero inferiore di funzioni alterate rispetto a quelle prodotte da mutazioni di componenti più a monte. In questo scenario è stato delineato il ruolo primario che il proto-o. ras svolge nella cascata di eventi cellulari che si innescano in risposta a stimoli di crescita. Molti fattori di crescita esercitano la loro funzione sulle cellule bersaglio solo dopo aver preso contatto con recettori specifici, che vanno incontro a dimerizzazione e autofosforilazione. Questi eventi determinano il reclutamento sul lato citoplasmatico della membrana di diverse proteine, definite adattatrici, che attraverso interazioni proteina-proteina vengono in contatto col prodotto del gene ras, fosforilandolo. Questa proteina fosforila a sua volta una serie di altre serin-treonin-chinasi che vanno infine ad attivare, fosforilandoli, fattori di trascrizione. Il repertoire di espressione genica di quella cellula viene a essere così modificato, iniziando una serie di eventi che porteranno alla trasformazione neoplastica della cellula.
Il concetto che le vie metaboliche possano ramificarsi a più livelli e che tali ramificazioni possano variare a seconda del tessuto è stato sostenuto per spiegare lo spettro di tessuto-specificità delle mutazioni a carico di proto-o. che, così modificati, si trasformano in oncogeni. Si è anche postulato che i vari proto-o. possano svolgere ruoli diversi nei diversi distretti cellulari, anche se nel caso dei tumori non poliposici del colon (HNPCC, Human Non Polyposis Colon Cancer) la funzione di riparo del DNA, che si ritiene alterata, venga svolta fisiologicamente in tutte le altre cellule dell'organismo. Pertanto è verosimile che vi siano altri eventi molecolari ancora non noti che determinano la tessuto-specificità dei tumori.
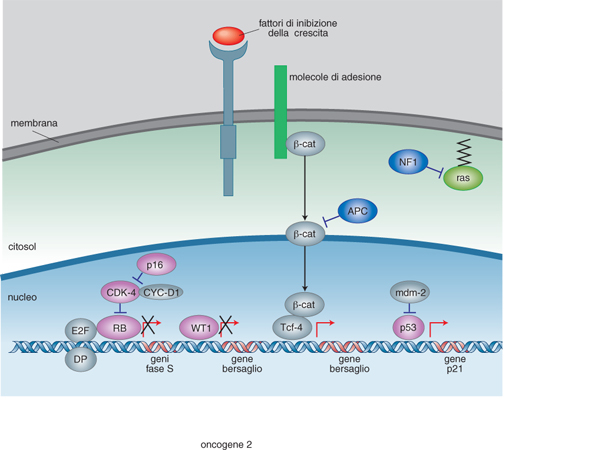
Lo studio dei tumori ereditari ha fatto anche progredire le nostre conoscenze sui geni soppressori di tumori, o oncosoppressori. Perché si possano instaurare questi tipi di tumori è necessario che siano mutati entrambi gli alleli del gene. Tali tumori possono essere sporadici, quando entrambi gli alleli del soggetto affetto vengono alterati per eventi mutazionali somatici indipendenti e diversi. Nei casi in cui sono ereditari, un cromosoma parentale porta una mutazione nel locus d'interesse, ma perché si instauri il tumore deve verificarsi un altro evento mutageno a livello somatico determinando l'alterazione della copia normale del gene. La componente ereditaria di questi tumori funziona, pertanto, con un meccanismo recessivo e il tumore si realizza solo quando entrambe le copie di quel gene sono alterate. I geni soppressori di tumori meglio caratterizzati sono quelli che codificano le proteine WT1, p53 e retinoblastoma, RB (fig. 2).
Mutazioni del gene WT1 sono responsabili del tumore di Wilms o nefroblastoma nel 5% dei casi ereditari e nel 5÷10% dei casi sporadici. Il tumore di Wilms è un tumore del rene dei bambini che si presenta approssimativamente in 1:10.000 nati nella maggior parte dei casi entro il 6° anno di età. È il tumore embrionale per eccellenza perché ricapitola lo sviluppo del suo organo corrispondente, il rene. Nelle forme ereditarie il meccanismo di trasmissione è autosomico recessivo, per cui entrambe le copie del gene responsabile devono essere alterate, come nel caso del retinoblastoma. Studi più recenti hanno però dimostrato che il 'modello di Knudson' dei due eventi mutageni a carico dello stesso gene oncosoppressore non è sufficiente a spiegare l'eziologia della malattia. Attualmente si ritiene che alla base della gran parte dei casi di questa sindrome tumorale vi sia un meccanismo genetico complesso che implica il coinvolgimento di almeno 10 geni soppressori. Pur non essendo WT1 il gene responsabile dell'inizio del processo tumorale, in tutti questi casi si ritiene che una sua alterata espressione possa contribuire al mantenimento dello stato tumorale. Il gene WT1 codifica una proteina zinc-finger (v. zinco, App. V) con diverse attività funzionali. È stato dimostrato che attraverso il motivo strutturale a dita di zinco, la proteina WT1 è in grado di legare molecole di RNA e di interagire con il DNA. Studi di espressione in vari sistemi cellulari in vitro hanno mostrato che il legame con il DNA a livello di sequenze di riconoscimento specifiche comporta sia attivazione che repressione trascrizionale. Infine, è stato riconosciuto un ruolo alla proteina WT1 nei fenomeni di morte programmata cellulare (apoptosi).
Mutazioni del gene RB che ne bloccano l'espressione sono responsabili del retinoblastoma, un tumore che colpisce la retina nella fanciullezza. RB è una proteina che va incontro a fosforilazione reversibile durante il ciclo cellulare. Nelle cellule quiescenti (G₀/G₁) la proteina non è fosforilata, lo diventa alla fine della fase G1 tramite i complessi cicline/chinasi ciclina-dipendente. Viene poi di nuovo defosforilata in fase M. Nella sua forma defosforilata, la proteina è capace di interagire specificamente con diverse proteine, tra cui i fattori di trascrizione della famiglia E2F. Questi ultimi attivano geni bersaglio i cui prodotti sono indispensabili per entrare nella fase S (o di sintesi o replicazione del DNA); l'interazione con RB inibisce la capacità transattivante di E2F per cui è stato suggerito che RB, reprimendo l'espressione di geni dipendenti da E2F, impedisce che le cellule entrino in fase S. Inoltre, il complesso RB-E2F è di per sé capace di attivare geni che ancora una volta impediscono alla cellula di entrare in fase S. In conclusione, la proteina RB blocca la proliferazione cellulare; tale attività deve essere soppressa perché la cellula possa progredire lungo il ciclo cellulare, attraverso una fosforilazione reversibile. L'espressione aumentata di RB impedisce la crescita cellulare, mentre cellule che non esprimono RB sono iperproliferanti.
Un altro importante gene soppressore di tumore è p53. Il prodotto genico è una fosfoproteina a localizzazione nucleare con peso molecolare di 53 kda (chilodalton), che funziona in forma di tetramero. Perché si produca tumore è necessario che entrambe le copie del gene siano delete o che un allele abbia una mutazione missense. In quest'ultimo caso, la specie mutante forma eterodimeri con quella normale, impedendone il funzionamento. Le forme mutanti si comportano perciò come mutanti dominanti negativi, cioè, pur interagendo con le forme normali, ne bloccano l'attività. Entrambe queste alterazioni sono state riscontrate nei più diversi tumori, suggerendo che p53 intervenga nei meccanismi generali di controllo della proliferazione cellulare, su cui in condizioni normali svolge un ruolo inibitorio. Normalmente, infatti, la cellula possiede bassi livelli di p53; sotto l'azione dei raggi ultravioletti o di qualunque altro stimolo capace di provocare danno al DNA, i suoi livelli aumentano e la cellula può andare incontro ad arresto della crescita o ad apoptosi (v. cellula, in questa Appendice). Il diverso destino dipende dalla fase del ciclo cellulare: se infatti la cellula si trova in una fase di G₁ precoce, p53 blocca la progressione nel ciclo attraverso l'inibizione dei complessi cicline/chinasi ciclina-dipendente. Questo blocco permette alla cellula di riparare i danni al DNA prodotti dallo stimolo, prima di entrare nella fase S di replicazione del DNA e, quindi, prima di incorporare la mutazione nel materiale genetico e di trasmetterla alla progenie. Se la cellula è già destinata alla divisione cellulare, allora p53 attiva il programma di morte cellulare, cioè innesca una serie di eventi molecolari che terminano con il collasso della cellula, la formazione di piccoli corpi picnotici e la frammentazione del DNA. L'esistenza di questi due possibili risultati finali può essere razionalizzata supponendo che il danno al DNA attivi la via della trasformazione cellulare e che il compito di p53 sia quello di proteggere l'organismo contro le sue conseguenze. Se è possibile, p53 attiva una serie di eventi atti a correggere gli errori sul DNA; se non è possibile, allora la cellula viene distrutta. Le fasi del ciclo in cui si trova la cellula e le sue caratteristiche determinano l'attivazione dell'una o dell'altra via possibili. Le tappe di questo processo e le molecole che vi partecipano non sono ancora del tutto note, anche se recentemente sono stati riconosciuti molti partner in base ai domini funzionali di p53 identificati e mappati sulla proteina. È stato possibile così identificare geni regolati da p53 che bloccano la divisione cellulare; i motivi che legano elementi regolatori di geni bersaglio; i motivi di interazione proteina-proteina, in particolare quelli di interazione con i fattori del complesso trascrizionale basale. Considerando il ruolo che p53 svolge in processi così importanti per la vita della cellula, non è sorprendente ritrovare forme mutate del gene associate a diverse forme tumorali, siano esse ereditarie o sporadiche. Mutazioni ereditarie di p53 predispongono principalmente a osteosarcomi, sarcomi dei tessuti molli, tumori cerebrali, leucemie e, nelle donne, tumori della mammella. Mutazioni somatiche del gene sono invece responsabili non solo di una consistente percentuale di tumori sporadici, ma anche dell'andamento più aggressivo di altri tumori. In prospettiva, il riconoscimento del coinvolgimento di p53 potrebbe essere utilizzato come mezzo diagnostico: il riscontro di una sua mutazione potrebbe rappresentare indicazione prognostica negativa e richiedere interventi terapeutici diversi e più incisivi.
Infine, lo studio e la delucidazione del funzionamento dei geni oncosoppressori ha fornito informazioni anche sui meccanismi con cui i virus tumorali a DNA sono in grado di trasformare le cellule di ospiti non naturali. In particolare, la capacità trasformante dei virus SV40 e Polyoma è da riportare a proteine virali che cooperano nell'indurre una cellula quiescente a proliferare. Il passaggio dalla fase G₁ a S è la norma nelle cellule permissive all'infezione virale perché consente la duplicazione del DNA virale. Se esso avviene in una cellula non permissiva vuol dire che i normali meccanismi di controllo del ciclo cellulare sono stati alterati in maniera irreversibile e trasmissibile alla progenie. Gli antigeni tumorali di SV40 e Polyoma si ritiene possano interagire con molecole regolatrici della crescita cellulare, interferendo o inibendo la loro azione. Il prodotto del gene p53, la proteina RB e gli altri membri della famiglia p107 e p130, come già detto, sono dei regolatori negativi della crescita cellulare. pRB e p130 sono capaci di bloccare la crescita interagendo con vari membri della famiglia E2F di fattori di trascrizione, reprimendo la trascrizione dei geni E2F-dipendenti. Nelle cellule che esprimono SV40 o Polyoma T antigen questi complessi sono distrutti, in quanto le proteine virali sono esse stesse capaci di legare questi fattori, rendendoli trascrizionalmente competenti. Si dereprime così una serie di geni responsabili della progressione della cellula lungo il ciclo cellulare, innescando una cascata di reazioni che porta alla loro trasformazione tumorale. Sono stati anche identificati i domini della proteina capaci di interagire con vari componenti dell'apparato cellulare responsabile della progressione nel ciclo cellulare. Lo studio dettagliato dei vari componenti che partecipano alla formazione di questi complessi e la delucidazione dei meccanismi di azione dovrebbe fornire ulteriori informazioni sulla trasformazione neoplastica da parte dei virus tumorali a DNA.
In conclusione, la ricerca sugli o., sia quella rivolta all'identificazione di nuovi membri, sia quella diretta al chiarimento dei meccanismi d'azione e degli effetti di mutazioni, ha ricevuto un considerevole sviluppo che si spera, in un futuro non lontano, possa produrre risultati positivi e applicativi nella pratica medica.
bibliografia
F.R. Fearon, Human cancer syndrome: clues to the origin of natural cancer, in Science, 1997, 278, pp. 1013-50.
B. Lewin, Genes VI, New York-Oxford-Tokyo 1997.
F.P. Perera, Environment and cancer: who are susceptible, in Science, 1997, 278, pp.1068-73.
E. Ponder , Genetic testing for cancer risk, in Science, 1997, 278, pp. 1050-54.
D. Sidransky, Nucleic acid based methods for the detection of cancer, in Science, 1997, 278, pp. 1054-58.