
neuropatie
neuropatie
Le neuropatie sono malattie del sistema nervoso periferico (SNP) con espressione clinica focale o diffusa. Possono essere acute o croniche e determinano alterazioni della forza, della sensibilità e delle funzioni autonomiche, molto spesso associate a dolore. Le neuropatie riconoscono molteplici eziologie che determinano un danno prevalente a carico delle guaine mieliniche ovvero degli assoni. Tra le forme acquisite prevalgono per incidenza quelle focali e tra le sistemiche quelle dismetaboliche e infettive. Le forme ereditarie sono determinate da mutazioni di molti geni e l’espressione fenotipica più frequente è la malattia di Charcot-Marie-Tooth. La diagnosi deve avvalersi dello studio elettrofisiologico e talora dell’esame del liquor e della biopsia del nervo. Una parte consistente delle neuropatie (25%) non ha una causa riconoscibile. La terapia è determinata dal riconoscimento dell’eziologia e prevede principalmente l’uso di trattamenti immunomodulanti, immunosoppressori, antidolorifici e vitaminici. [➔ cervello, struttura e funzione; Guillain-Barré, sindrome di; neurodegenerazione; sclerosi multipla; tunnel carpale, sindrome del; vasculopatie]
Le n. sono malattie dell’SNP che possono interessare un solo tronco nervoso (mononeuropatie), più tronchi nervosi con distribuzione casuale (multineuropatie) o tutti i tronchi nervosi degli arti, iniziando simmetricamente dai segmenti più distali (polineuropatie). Il decorso clinico può essere acuto o cronico, e si caratterizza per la diminuzione della forza (ipostenia) e del volume muscolare (ipotrofia), per le alterazioni delle sensibilità (ipoestesie, parestesie e disestesie) e per le modificazioni delle funzioni autonomiche. L’agente causale danneggia selettivamente le cellule di Schwann o le guaine mieliniche, determinando demielinizzazione e relativo risparmio degli assoni, oppure degenerazione degli assoni mediante azione lesiva sul corpo cellulare o sull’assolemma (sottile membrana che avvolge l’assone). In entrambe le condizioni patologiche è possibile che siano interessate tutte le fibre nervose o prevalentemente alcune di esse. Dalla diversa combinazione degli eventi patologici e delle fibre principalmente interessate dalla malattia scaturisce la variabilità del quadro clinico. Le n. possono essere determinate da molteplici cause, essere acquisite o geneticamente determinate e globalmente hanno nella popolazione generale una prevalenza di circa il 3%. La prevalenza aumenta fino all’8% se si considera la popolazione con età superiore ai 55 anni. Nel mondo occidentale la forma di n. più frequente è quella diabetica, nell’America Meridionale, in Asia e in Africa la causa più frequente è la lebbra. Altre malattie dismetaboliche e infettive, le vasculopatie, i farmaci, le sostanze tossiche e le neoplasie contribuiscono differentemente all’eziologia. Le n. disimmuni e le forme geneticamente determinate prevalgono nelle forme croniche. L’ipotesi clinica di n. deve essere confermata dallo studio elettrofisiologico, che è il metodo più specifico e sensibile per valutarne l’estensione (mono-, multi- o polineuropatia) e per stabilirne la natura demielinizzante o degenerativo-assonale.
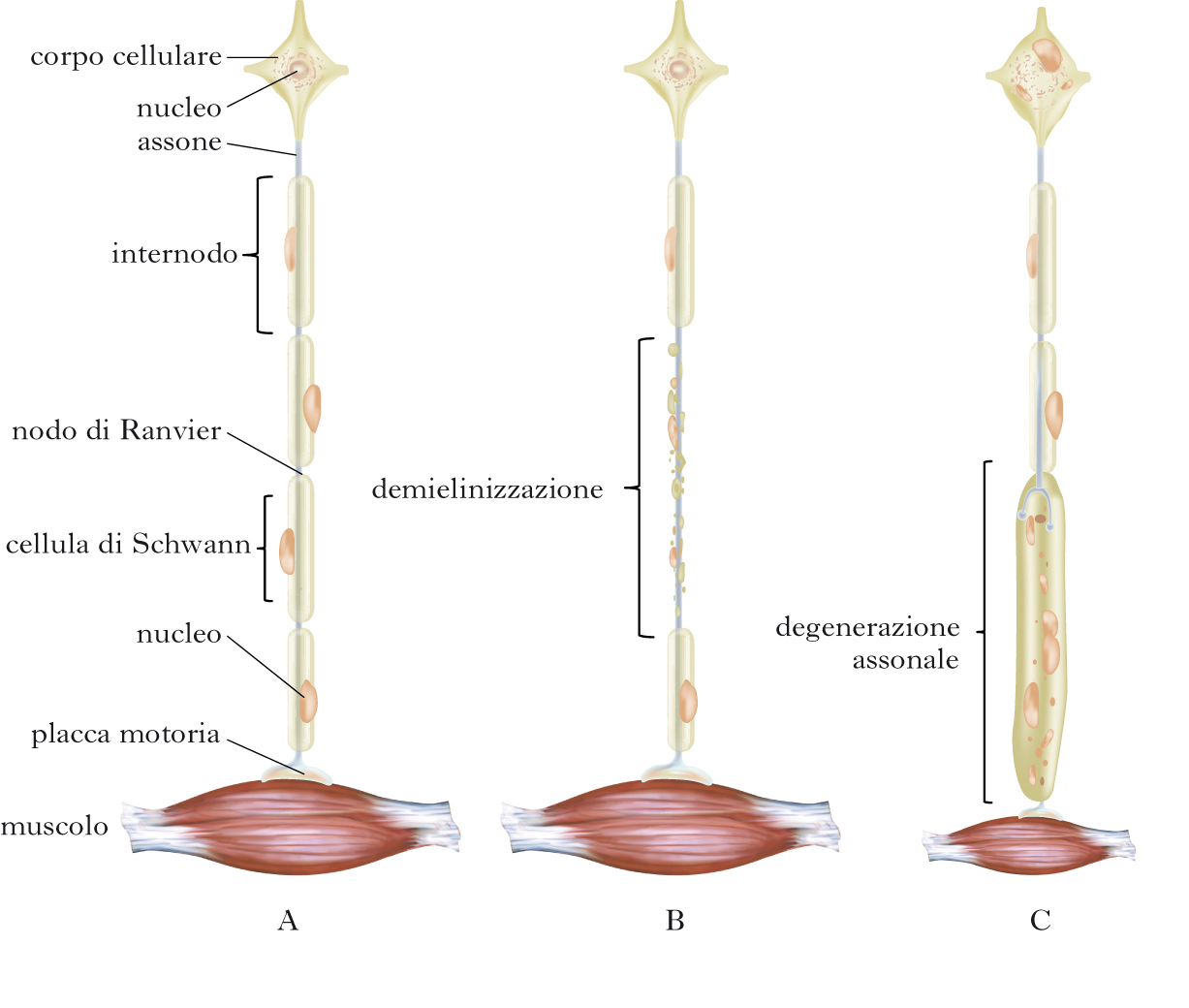
Mononeuropatie
L’interessamento isolato di un tronco nervoso è generalmente dovuto a un trauma locale, a compressione-intrappolamento del nervo in una specifica regione anatomica o infiltrazione neoplastica, e rappresenta la più circoscritta patologia dell’SNP. Le mononeuropatie degli arti superiori e in partic. quelle che interessano il nervo mediano al polso (➔ tunnel carpale, sindrome del) e l’ulnare al gomito o al polso sono le più frequenti. Tuttavia, altri nervi degli arti superiori (nervo radiale e ascellare), del tronco (sovrascapolare e toracico lungo) e degli arti inferiori (nervo femorale, femorocutaneo laterale, otturatorio, tibiale posteriore e peroneo) possono essere coinvolti in n. focali. Tutte le mononeuropatie sono caratterizzate da un deficit funzionale, motorio e sensitivo, limitato a un territorio innervato esclusivamente da un tronco nervoso, presentando quindi un quadro clinico specifico che dipende dal nervo interessato. Queste peculiarità rendono relativamente semplice la diagnosi di mononeuropatia. La decisione sul trattamento conservativo o chirurgico dipende dalla valutazione clinica di transitorietà, cronicità o peggioramento dei deficit neurologici e dai dati elettrofisiologici. Questi infatti devono confermare la focalità della lesione ed evidenziare la presenza di demielinizzazione o di degenerazione assonale.
Multineuropatie
Si caratterizzano sul piano clinico per l’interessamento, simultaneo o progressivo, di più nervi periferici non contigui, con distribuzione multifocale e in genere con evoluzione rapida. In alcune occasioni le lesioni multiple si accumulano nel tempo fino a determinare una condizione di patologia simmetrica e diffusa, tale da rendere difficile la differenziazione da una polineuropatia. In questi casi è indispensabile ricostruire accuratamente l’esordio della sintomatologia e la sua evoluzione. Una corretta e precoce diagnosi di multineuropatia è essenziale in quanto la maggior parte di esse è sostenuta da processi vasculitici sistemici, che fanno parte di malattie del connettivo. Altre malattie sistemiche che possono indurre una multineuropatia sono la sarcoidosi, l’amiloidosi, i linfomi, i carcinomi e l’AIDS. La malattia diabetica può determinare una particolare forma di microvasculite focale che interessa abitualmente il plesso lombosacrale e si manifesta con acuto e persistente dolore, ipotrofia dei muscoli prossimali dell’arto inferiore e conseguente ipostenia (amiotrofia diabetica). In alcuni casi di multineuropatie, nei quali la diagnosi rimane incerta, può essere necessario ricorrere alla biopsia di un nervo periferico (surale o peroneo superficiale). Accertata la vasculite, il trattamento si fonda sulla terapia immunosoppressiva con steroidi, ciclofosfamide o altri farmaci analoghi.
La n. motoria multifocale (MMN, Multifocal Motor Neuropathy) è una sindrome piuttosto rara che si manifesta clinicamente come una multineuropatia, ma con esclusivo interessamento delle fibre motorie e con blocchi focali della conduzione nervosa. Essa non è sostenuta da processi vasculitici e la patogenesi consiste nell’aggressione di specifici gangliosidi di membrana a livello dei nodi di Ranvier, da parte di determinati anticorpi (GM1). La sintomatologia consiste essenzialmente in una paresi o, più raramente, una plegia con decorso cronico di più muscoli innervati da almeno due nervi periferici. La MMN può essere curata con infusione endovena di immunoglobuline G, mentre non è sensibile ai trattamenti con steroidi. Un quadro clinico di multineuropatia con blocchi focali della conduzione può essere anche sostenuto dalla n. ereditaria con suscettibilità alla paralisi da compressione (HNPP, Hereditary Neuropathy with liability to Pressure Palsies).
Polineuropatie
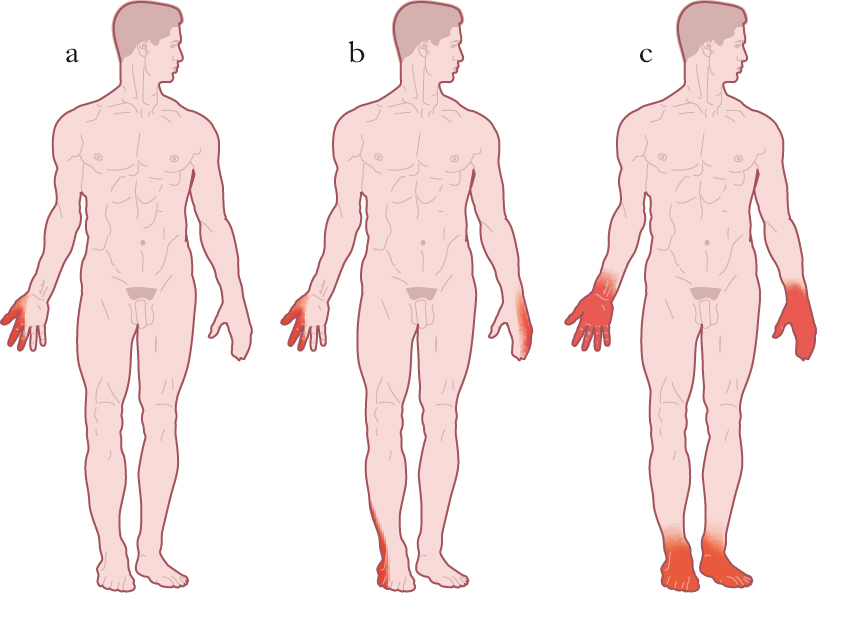
Le polineuropatie si presentano con un interessamento simmetrico dei nervi periferici che inizia nei segmenti più distali degli arti inferiori, si diffonde in senso prossimale, e coinvolge poi anche le mani e gli arti superiori. Si ritiene che questa presentazione della sintomatologia sia dovuta al coinvolgimento delle fibre nervose secondo la modalità lunghezza-dipendente. In pratica, più lunghe e più distanti dal corpo cellulare sono le fibre nervose, più sensibili al danno patogeno diventano i loro segmenti distali. Questa selettiva vulnerabilità può dipendere da difficoltà per il corpo cellulare di sintetizzare proteine strutturali, da alterazioni del trasporto assonale o da deficit distrettuali del metabolismo energetico. Le manifestazioni cliniche di queste n. sono molto variabili, ma abitualmente la sintomatologia insorge con un disturbo sensitivo spiacevole spontaneo (parestesie), o indotto da stimoli abitualmente innocui (disestesie), alle dita e alle piante dei piedi, e consistente in senso di formicolio, punture di spillo, senso di costrizione o intorpidimento. Successivamente, parestesie e disestesie si diffondono lungo le gambe con distribuzione a calza e si associano a ipoestesia, ipostenia dei muscoli estensori delle dita e del piede e scomparsa del riflesso achilleo. In questa fase compaiono anche parestesie e disestesie alle mani con distribuzione a guanto e difficoltà nella deambulazione, dovute alla ipostenia e alla riduzione della sensibilità propriocettiva (senso di posizione degli arti), con conseguente incertezza nella marcia. Se la malattia progredisce ulteriormente, si può avere diffusione dell’ipoestesia e dell’ipostenia anche nei segmenti più prossimali degli arti e sull’addome, incapacità a deambulare e scomparsa di tutti i riflessi osteotendinei. È frequente anche una sintomatologia dolorosa con iperalgesia (stimolo doloroso avvertito in modo eccessivo) o allodinia (stimolo non nocicettivo, avvertito come doloroso). Infine, possono manifestarsi sintomi e segni di disautonomia per il coinvolgimento delle fibre simpatiche e parasimpatiche. Le alterazioni della sudorazione (ipo- o anidrosi) e dei riflessi pupillari, i disturbi dell’erezione, le irregolarità dell’alvo e l’ipotensione ortostatica sono le manifestazioni più frequenti. I sintomi e i segni descritti possono combinarsi in maniera molto variabile a seconda della funzione delle fibre nervose prevalentemente o esclusivamente interessate dall’evento patologico. È possibile dunque che le polineuropatie siano motorie, sensitive, disautonomiche o miste. Le differenti sensazioni parestesiche/disestesiche, l’ipoe;stesia globale o parziale e la presenza di dolore dipendono inoltre dal calibro delle fibre sensitive coinvolte. L’ipoestesia globale a calza e guanto con sintomi disautonomici, l’ipotrofia e l’ipostenia muscolare distale con l’assenza dei riflessi osteotendinei rappresentano l’esemplificazione di un quadro clinico conclamato di polineuropatia mista. Le polineuropatie possono essere acquisite o geneticamente determinate. Per le forme acquisite è possibile riconoscere numerose varianti, tra le quali le più rilevanti sul piano epidemiologico sono le polineuropatie infiammatorie disimmuni, quelle associate a malattie sistemiche o a presenza di proteine monoclonali, quelle di origine tossica o infettiva. Va comunque ricordato che ca. nel 25% delle polineuropatie non è possibile individuare un’eziologia certa.
Polineuropatie acquisite
Polineuropatie infiammatorie disimmuni. Sono caratterizzate da un coinvolgimento contemporaneo delle radici e dei nervi periferici, da un processo patologico di tipo prevalentemente demielinizzante e da un decorso clinico acuto o cronico. La forma acuta è rappresentata dalla sindrome di Guillain-Barré (➔) che raggiunge l’acme della sintomatologia nell’arco di pochi giorni.
Poliradicoloneuropatia cronica infiammatoria demielinizzante (CIDP). Questa polineuropatia ha un decorso clinico molto più protratto, è raramente preceduta da un evento infettivo e può avere una evoluzione progressiva (60% dei pazienti), ovvero un andamento recidivante con guarigione tra un episodio e il successivo. La CIDP si manifesta in ogni epoca della vita ma la prevalenza aumenta con l’età fino a un massimo nella sesta e settima decade e colpisce più frequentemente i maschi. Le manifestazioni cliniche sono molto variabili, ma nella maggior parte dei pazienti la sintomatologia si sviluppa simmetricamente, coinvolge il versante sia motorio sia sensitivo, sebbene l’ipostenia sia abitualmente prevalente e interessi sia gli arti superiori sia quelli inferiori, con più spiccato coinvolgimento di questi ultimi. L’assenza dei riflessi osteotendinei è praticamente costante. Rari sono l’interessamento dei nervi cranici e la presenza di dolore, mentre mai si osserva una sintomatologia disautonomica. In una minoranza di pazienti è possibile che si manifesti un quadro clinico asimmetrico, che ricorda una multineuropatia, ma che ha le medesime caratteristiche patologiche ed elettrofisiologiche della forma simmetrica. La diagnosi clinica di CIDP può essere avanzata quando la sintomatologia dura da almeno due mesi, l’ipostenia coinvolge contemporaneamente muscoli distali e prossimali, nel liquor vi è un aumento abnorme di proteine e le indagini elettrofisiologiche dimostrano la presenza di evidenti segni di demielinizzazione con rallentamento disomogeneo della conduzione ed eventuali blocchi della stessa. Nei casi nei quali non si riscontra l’aumento delle proteine nel liquor, la biopsia del nervo può essere utile per confermare la diagnosi e per escludere altre cause di neuropatia. È importante la precocità della diagnosi perché la CIDP risponde ai trattamenti con gli steroidi, la plasmaferesi o l’infusione endovena di immunoglobluline G. Tuttavia, nonostante la maggior parte dei pazienti presenti un evidente miglioramento iniziale con la terapia, il decorso successivo non può essere predetto e il tasso di recidive è piuttosto elevato. Inoltre molti pazienti presentano un’evoluzione dalla forma remittente a quella cronica progressiva.
Polineuropatie associate a malattie sistemiche. Il prototipo di queste n. è rappresentato dalla forma cronica sensorimotoria e disautonomica che può svilupparsi nel corso della malattia diabetica e che interessa ca. il 50% dei pazienti dopo 5 anni di malattia. Il processo patologico, prevalentemente di tipo degenerativo assonale, coinvolge con modalità lunghezza-dipendente le fibre mieliniche − di grande e di piccolo calibro − e amieliniche. L’esordio della sintomatologia è insidioso con disturbi prevalentemente sensitivi, localizzati alle estremità degli arti inferiori. Il paziente tipicamente lamenta intorpidimento dei piedi, sensazione di camminare su un tappeto di spilli o su cocci di vetro, e ha sensazione di instabilità. Clinicamente si osservano ipoestesia globale a calza, con occasionale allodinia, assenza dei riflessi achillei, cute secca e sottile con riduzione dei peli. Nel tempo, la sintomatologia interessa anche gli arti superiori e coinvolge anche le fibre motorie con ipostenia e ipotrofia. La grave riduzione della sensibilità nei piedi, la microangiopatia e le deformità articolari inducono la manifestazione di ulcere cutanee con necessità di amputazioni che interessano una percentuale non trascurabile di pazienti. Una variante della polineuropatia su descritta è rappresentata dalla polineuropatia delle piccole fibre che coinvolge esclusivamente o prevalentemente le fibre di piccolo calibro. In questi casi i pazienti lamentano importanti disturbi sensitivi con parestesie, disestesie e allodinia. Talora i fenomeni allodinici sono suscitati anche dal contatto con le lenzuola, determinando impossibilità al riposo notturno. Questa variante di polineuropatia è associata molto spesso alla condizione prediabetica di intolleranza ai glucidi. I meccanismi patogenetici delle n. diabetiche sono molteplici, quelli maggiormente accreditati sono lo stress ossidativo cronico e le alterazioni della microcircolazione di natura metabolica e infiammatoria autoimmune. Le neoplasie maligne possono indurre la manifestazione in modo acuto o subacuto di una polineuropatia prevalentemente sensitiva. La sintomatologia neuropatica può precedere di mesi o anni il riconoscimento della neoplasia, spesso rappresentata da un carcinoma polmonare a piccole cellule. Il quadro clinico è dominato dalla sintomatologia sensitiva, molto spesso anche dolorosa, con coinvolgimento peculiare della sensibilità propriocettiva e conseguente atassia della marcia e incoordinazione degli arti. La quasi totalità dei pazienti presenta nel siero e nel liquor positività per anticorpi antinucleari e antineuronali, denominati Hu. Le carenze vitaminiche, in partic. di quelle del gruppo B, spesso secondarie ad assunzione eccessiva di alcol, possono determinare l’insorgenza insidiosa e cronicamente progressiva di una polineuropatia sensorimotoria, caratterizzata da degenerazione assonale con modalità lunghezza-dipendente. L’adegua;mento della dieta e la supplementazione vitaminica possono indurre una lenta rigenerazione assonale.
Polineuropatia associata a proteina monoclonale. In una significativa percentuale (10%) di pazienti della sesta÷settima decade che presentano un esordio molto insidioso e una progressione lenta di una polineuropatia sensorimotoria distale, si osserva la presenza di una proteina monoclonale (proteina M). L’individuazione di una polineuropatia associata a proteina monoclonale può determinare la scoperta di una amiloidosi sistemica primaria, di un mieloma multiplo, di un linfoma o di altra malattia linfopro;liferativa maligna, ricadendo quindi nel caso delle polineuropatie associate a malattie sistemiche. In molti pazienti non è tuttavia possibile individuare alcuna patologia associata: questi casi vengono definiti come gammopatie monoclonali di incerto significato (MGUS) e vanno monitorati. Spesso il quadro clinico ed elettrofisiologico è molto simile a quello di una CIDP con maggiore incidenza di atassia e tremore. I casi più severi devono essere trattati con plasmaferesi o immunosoppressori.
Polineuropatie tossiche. Queste polineuropatie possono essere indotte da numerose sostanze di origine industriale e ambientale e da molti farmaci (➔ nevriti). Solitamente la sospensione del neurotossico fa regredire la sintomatologia. I farmaci antineoplastici rappresentano attualmente la causa principale di polineuropatie iatrogene, che sono abitualmente sensitive e dose dipendenti. Anche alcuni antimicrobici (isoniazide, cloramfenicolo, nitrofurantoina), gli antiretrovirali e l’amiodarone possono indurre una polineuropatia dopo un periodo prolungato di assunzione.
Polineuropatie infettive. Durante l’infezione da HIV si possono osservare frequentemente polineuropatie infettive, che assumono caratteristiche e decorso differenti a seconda dello stadio di malattia. Durante la sieroconversione le n. assumono caratteristiche indistinguibili dalla sindrome di Guillain-Barré, mentre negli stadi tardivi sintomatici la polineuropatia è a lenta evoluzione, distale, simmetri;ca e prevalentemente sensitiva, spesso con cospicua componente dolorosa.
Polineuropatie ereditarie
Le polineuropatie ereditarie (PE) riguardano 1 individuo su 2.500, sono pertanto una delle malattie ereditarie neurologiche più frequenti e costituiscono un capitolo complesso ed eterogeneo. Possono essere trasmesse con modalità autosomica dominante o recessiva, oppure essere legate al cromosoma X. Il fenotipo è estremamente variabile, con esordio abitualmente insidioso, nella prima o nella seconda decade, il decorso è cronico con possibilità di sviluppare una disabilità permanente. La classificazione delle PE prevede la suddivisione in n. ereditarie sensorimotorie (HMSN, Hereditary Motor and Sensory Neuropathy), diffusamente definite con l’eponimo di malattia di Charcot-Marie-Tooth, n. ereditarie sensoriautonomiche (HSAN, Hereditary Sensory and Autonomic Neuropathy) e n. ereditarie motorie (HMN, Hereditary Motor Neuropathy).
La malattia di Charcot-Marie-Tooth (CMT) può senz’altro essere considerata il prototipo delle PE per la sua elevata frequenza, per la variabilità fenotipica e per la complessità dei meccanismi patogenetici. Molte famiglie con CMT dominante presentano quadri elettrofisiologici di demielinizzazione, generati da mutazioni in geni espressi dalle cellule di Schwann che formano la mielina periferica (CMT1). Altre famiglie, meno numerose, esprimono aspetti di degenerazione assonale, e hanno mutazioni su geni espressi dai neuroni (CMT2). Attualmente sono noti molti geni che possono indurre un fenotipo CMT con ipostenia e atrofia muscolare distale, riduzione delle sensibilità e deformità dei piedi (piedi cavi). Nonostante il grande sviluppo delle conoscenze sui meccanismi molecolari, non si dispone ancora di terapie sicuramente efficaci. Sono importanti il monitoraggio delle possibili deformità ossee e la pratica costante di terapia fisica riabilitativa. Lucio Santoro
Neuropatie da farmaci
La moltiplicazione e la diffusione di molecole farmacologicamente attive ha contribuito, negli ultimi decenni del 20° sec., all’elevata incidenza delle neuropatie da farmaci e al chiarimento di alcuni fra i meccanismi patogenetici, sebbene in alcuni casi si possa solamente prendere atto di una correlazione evidente, del tipo causa-effetto, fra la somministrazione di una sostanza e la comparsa di una patologia neuritica. La neuropatia iatrogena è da considerarsi in molti casi come rischio accettato, dal momento che i farmaci che la inducono possono essere salvavita, come quelli antitumorali; in altri casi (antibiotici neurotossici) l’uso è da riservare solo laddove essi si dimostrino indispensabili senza alternative terapeutiche, e comunque è indicato lo stretto monitoraggio di segni e sintomi di neuropatia incipiente, spec. a carico dei nervi cranici.
Neuropatie del nervo acustico da antibiotici amminoglicosidici
Già negli anni Cinquanta del 20° sec. era evidente che la somministrazione intramuscolare di streptomicina (un antibiotico della classe degli amminoglicosidi molto usato per la terapia di infezioni tubercolari e altre infezioni croniche batteriche) poteva provocare sordità e alterazioni della funzionalità vestibolare: fu forse la prima evidenza clinica di una neurite cranica (dell’VIII paio di nervi cranici) da farmaco. A lungo tuttavia non si conobbe il meccanismo patogenetico di tale iatrogenicità: attorno al 1980 si scopri l’accumulo patologico degli amminoglicosidi nella perilinfa e nell’endolinfa dell’orecchio interno, proporzionale alla concentrazione ematica, e successivamente si evidenziò che, per tutti i farmaci di questa classe, anche una sola dose (per es., di tobramicina) è in grado di produrre effetti ototossici, e che tale patologia è solitamente irreversibile a causa della distruzione delle cellule sensitive cocleari e vestibolari. Ciò impedisce al sistema uditivo di generare potenziali d’azione in risposta ai suoni e ai movimenti. L’aumento del dosaggio e la somministrazione prolungata, ma soprattutto la ripetizione di cicli terapeutici, provocano inoltre la progressione a ritroso del danno per tutto il nervo acustico. Anche applicazioni intrauricolari (in caso di otiti con perforazione timpanica) di amminoglicosidi, polimixina-colistina, cloramfenicolo, possono determinare alterazioni cocleovestibolari. L’ipotesi patogenetica più probabile è che gli amminoglicosidi alterino dapprima la concentrazione di ioni nei fluidi del labirinto e secondariamente interagiscano con i fosfolipidi di membrana del nervo.
Neurite ottica
Questa infiammazione con danno anatomofunzionale del nervo ottico si riscontra talvolta dopo somministrazione di etambutolo a dosaggi elevati e trattamenti prolungati, spec. in pazienti con insufficienza renale ed epatica, diabete, etilismo; eccezionalmente compare anche con isoniazide (associata all’etambutolo nel trattamento antitubercolare) e cloramfenicolo; quest’ultimo antibiotico può provocare, nei pazienti con fibrosi cistica e nei neonati, perdita simmetrica delle cellule gangliari della retina e atrofia delle fibre del nervo ottico, potendosi accompagnare, se pur raramente, a una neuropatia periferica con parestesie. Tra i farmaci più recenti (2001) sono soprattutto l’etanercept e altri inibitori del TNF-α (Tumor Necrosis Factor-α ) a essere chiamati in causa, per patologie oculari di tipo demielinizzante, quali la neurite ottica, e di tipo infiammatorio (uveiti). Non è stato ancora chiarito il meccanismo alla base di queste reazioni avverse: questi farmaci potrebbero esacerbare le malattie demielinizzanti inibendo l’apoptosi di cellule T potenzialmente autoreattive e innescando, quindi, una risposta autoimmunitaria contro la mielina a livello del sistema nervoso centrale. Un diverso meccanismo patogenetico, di tipo ischemico, è imputabile alla nevrite ottica da sildenafil (Viagra) e da amiodarone: essi sono ritenuti responsabili del 42% di tutti i casi di neuropatia ottica ischemica.
Neuropatie da alcaloidi della Vinca e da altri farmaci antineoplastici
Alcuni alcaloidi isolati da Vinca rosea e usati come antineoplastici hanno spiccata tossicità per le strutture nervose e in partic. per quelle periferiche. Le nevriti si manifestano con parestesia, dolori di tipo folgorante e difetti trofici. La vincristina può causare anche neuropatie a livello del sistema nervoso autonomo, con costipazione, ileo paralitico, dolore addominale, ritenzione urinaria e ipotensione ortostatica. Un’altra classe di farmaci con elevata tossicità è quella delle molecole stabilizzanti i microtubuli, tra cui i taxani (come il paclitaxel e il docetaxel): essi possono causare l’insorgenza di una neuropatia grave, che dipende dalla dose per ciclo di trattamento e dalla durata dell’infusione, ma che generalmente si risolve in maniera graduale dopo l’interruzione del trattamento. La ragione di questi effetti risiede nel meccanismo d’azione: interferendo con i microtubuli delle strutture nervose (il paclitaxel promuove l’aggregazione dei polimeri di tubulina, mentre la vincristina ne promuove la depolimerizzazione), questi farmaci impediscono il trasporto di proteine essenziali nei granuli secretori a livello delle cellule nervose. Il metotrexato, che induce invece il rilascio di adenosina nel sistema nervoso centrale e inibisce la sintesi delle purine, può ugualmente provocare neurotossicita a livello dei nervi cranici con deficit dei muscoli oculari, ptosi, paralisi facciale e laringea, e deficit dell’acuità visiva da compromissione del nervo ottico. Gli antineoplastici suddetti possono dare anche neurotossicita periferica di tipo sensoriale, con deficit sensitivo che si distribuisce ‘a guanto’ e ‘a calzino’. Simile la neuropatia da cisplatino e da oxaliplatino (utilizzato nel trattamento dei tumori gastrointestinali), con la perdita della propriocezione e della sensibilità vibratoria e iporeflessia; è di solito reversibile, ma il recupero avviene lentamente, nel giro di molti mesi dalla sospensione del farmaco.