neurofibromatosi
neurofibromatosi
Termine che accomuna due malattie eredo-familiari (n. di tipo 1 e n. di tipo 2), caratterizzate dalla tendenza a sviluppare tumori dei nervi periferici e di altri organi e da ereditarietà autosomica dominante. Le due malattie differiscono per manifestazione clinica, incidenza nella popolazione, causa genetica.
Neurofibromatosi di tipo 1 (NF1)
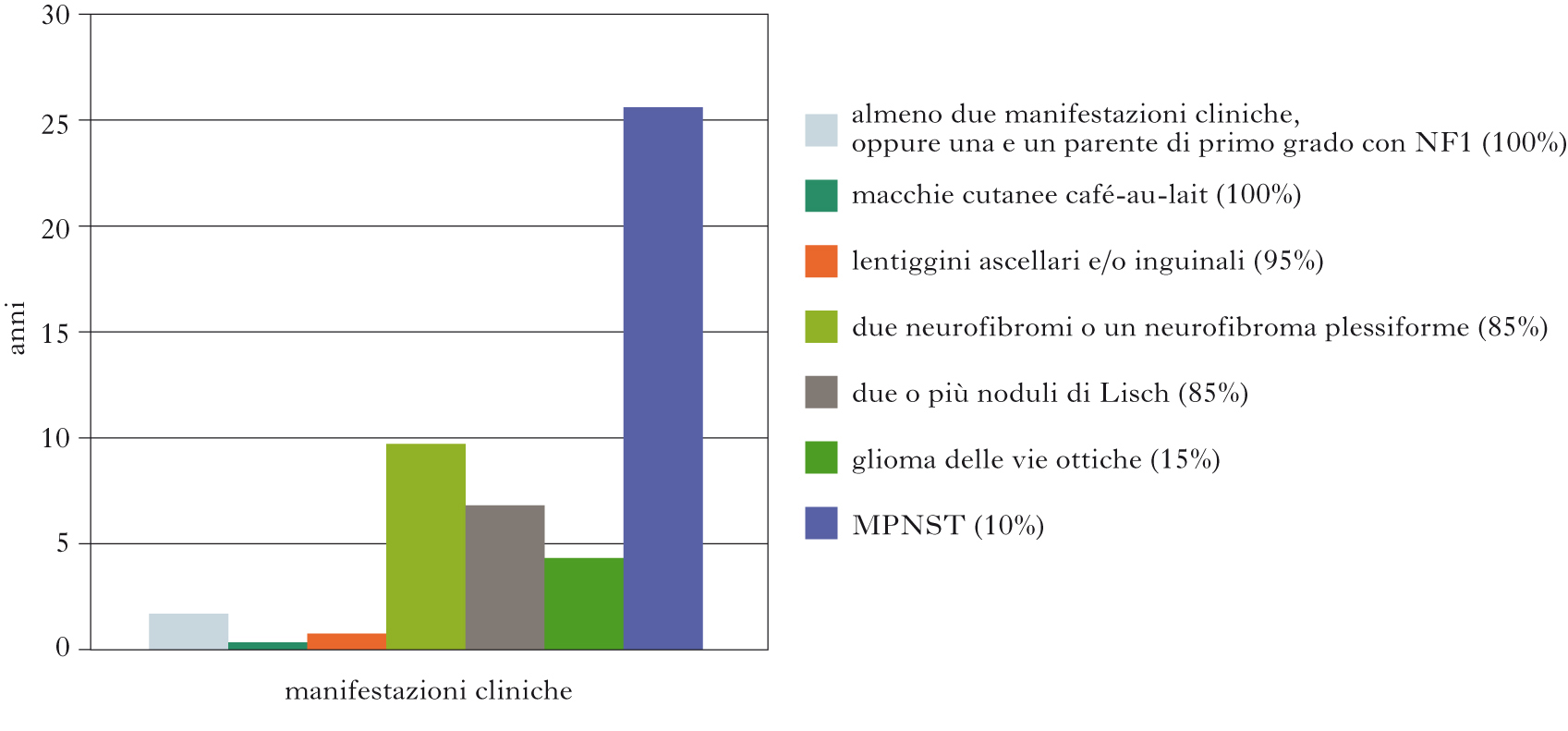
È nota anche come malattia di Von Recklinghausen, ha una prevalenza di 1:3.000, con frequenza più alta in popolazioni arabe e araboisraeliane. La penetranza è completa (ossia la mutazione si esprime sempre), ma l’espressione fenotipica è variabile; è riportato fino al 50% di mutazioni spontanee. Numerosi sono i segni clinici caratteristici della NF1. Le macchie cutanee color caffelatte (café-au-lait spots) sono presenti alla nascita e si ingrandiscono dopo la pubertà; altre lesioni cutanee sono lentiggini all’ascella e all’inguine. I neurofibromi cutanei, che sono tumori delle cellule delle guaine dei nervi (cellule di Schwann e fibroblasti), possono insorgere nei primi due anni di vita e talora estendersi in numerose e vaste are del corpo al punto da causare deturpamento della figura (neurofibroma plessiforme). Il neurofibroma plessiforme può andare incontro a trasformazione maligna diventando il tumore maligno delle guaine del nervo periferico, noto con l’acronimo MPNST (Malignant Peripheral Nerve Sheath Tumor). In alcuni casi la forma maligna è presente ab initio. Altro tumore tipico della NF1 è il glioma (➔) del nervo ottico, talora bilaterale, con scarsa propensione alla crescita invasiva; altri tumori intracranici (astrocitoma, glioblastoma) sono rari. Sono presenti quasi sempre nei pazienti adulti amartomi (piccole masse congenite similtumorali) dell’iride, noti come noduli di Lisch, e displasie ossee dello sfenoide e delle vertebre, che possono causare deformità dell’orbita e scoliosi. La presenza di almeno due tra i suddetti segni, oppure anche solo di uno accompagnato dalla presenza di un parente di primo grado con NF1, è sufficiente per la diagnosi. Altre manifestazioni che tendono a comparire nei pazienti con NF1 sono macrocefalia, deficit intellettivi, epilessia, idrocefalo e tumori del sistema ematopoietico e neuroendocrino. La NF1 è dovuta a mutazioni (ne sono state identificate più di 300) del gene NF1 situato sul cromosoma 17q11.2. Il prodotto di questo gene è una proteina detta neurofibromina; la sua funzione non è al momento precisata, ma agirebbe da oncosoppressore in quanto la sua perdita sembra responsabile di attivazione di vie promuoventi la mitosi. La NF1 è una malattia progressiva; cause di morte precoce sono il neurofibroma plessiforme massivo del mediastino, l’astrocitoma, il neurofibroma cervicale paraspinale e il MPNST. Il rischio di MPNST in un paziente con NF1 è del 3% circa. Questo tumore è raro nella prima decade di vita, si manifesta nell’adolescenza o in età giovane-adulta e ha una prognosi infausta per la tendenza all’invasione e alle metastasi.
Neurofibromatosi tipo 2 (NF2)
Questa n. è caratterizzata da lesioni tumorali o displastiche che colpiscono il sistema nervoso. La lesione più tipica è lo schwannoma (➔) bilaterale del nervo vestibolare, la cui comparsa in età inferiore a 30 anni è sufficiente a porre diagnosi certa di NF2. L’incidenza riportata (dati 2005) è di 1:25.000 neonati. Circa metà dei casi non ha storia familiare ed è dovuta a nuove mutazioni germinali. Altre manifestazioni della malattia che, se presenti in pazienti con storia familiare, consentono la diagnosi certa di NF2 sono il meningioma, lo schwannoma singolo, il glioma e l’opacità posteriore subcapsulare del cristallino. In assenza di familiarità, la diagnosi di probabile NF2 si formula quando una delle manifestazioni aggiuntive si presenta insieme a meningiomi multipli o schwannoma vestibolare in età inferiore a 30 anni. Lo schwannoma vestibolare della NF2 si differenzia da quello sporadico perché insorge in età più precoce (terza decade) ed è bilaterale ca. entro una decade. I meningiomi multipli sono un tratto tipico della malattia, specialmente nei pazienti pediatrici. Circa l’80% dei tumori gliali nella NF2 è localizzato nel midollo spinale, a differenza dei gliomi in generale, che sono prevalentemente cerebrali e consistono per lo più in ependimomi. Il gene della NF2 si trova sul cromosoma 22q12. Le mutazioni somatiche e germinali identificate sostengono l’ipotesi che il prodotto del gene abbia funzione di gene oncosoppressore. Il prodotto proteico del gene NF2 è una proteina detta merlina, o anche schwannomina, la cui funzione non è nota con precisione. È possibile che agisca come oncosoppressore tramite regolazione della trasmissione di segnali dall’ambiente extracellulare a vie molecolari cellulari implicate nella proliferazione. Il decorso clinico dei pazienti con NF2 varia molto tra le diverse famiglie e anche all’interno delle famiglie stesse. Talora si ha un esordio precoce con un grosso carico di tumori, talora un esordio tardivo con solo schwannomi vestibolari. Il decorso più grave sembra associato alla ereditarietà matrilineare e alla anticipazione genica, ossia l’età di insorgenza e la gravità di espressione sono più precoci in ogni generazione successiva. Sono segnalate alcune correlazioni tra genotipo e fenotipo: la posizione della mutazione nel gene, così come alcuni tipi di mutazione, risultano corrispondere a un fenotipo clinico più grave. Un aumentato rischio di morte nei pazienti con NF2 è associato anche a esordio precoce di malattia e a presenza di meningiomi intracranici.