
neurodegenerazione
neurodegenerazione
Con l’aumento dell’età media della popolazione, più frequenti si fanno le malattie neurodegenerative. Pur se assai diverse dal punto di vista clinico, queste patologie hanno spesso una caratteristica comune, ossia la presenza di aggregati proteici tossici per cellule e neuroni. I ricercatori stanno cominciando a comprenderne i meccanismi di formazione, aprendo così la strada a nuove strategie terapeutiche. [➔ cervello, malattie genetiche del; demenza; imaging cerebrale, diagnosi e ricerca; movimento, disturbi del; neurofarmacologia]
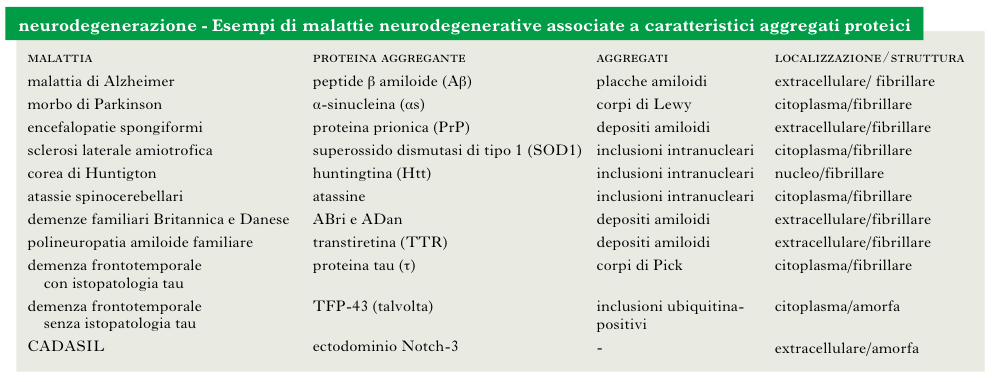
Il costante aumento dell’aspettativa di vita nei Paesi occidentali si accompagna inevitabilmente all’insorgere di patologie legate all’invecchiamento (➔ invecchiamento cerebrale). Tra queste, le malattie legate alla n. rappresentano un problema di crescente importanza per la nostra società. L’aspettativa di vita media è funzione delle condizioni socioambientali di un Paese: attualmente varia tra i 35 anni circa nello Swaziland e gli 80 in Giappone, Francia e Italia. La durata massima sembra invece determinata geneticamente: per un uomo, si calcola intorno a 120 anni. Perché mai? L’invecchiamento e la morte dei singoli individui sono essenziali all’evoluzione di qualsiasi specie. Si deve lasciare spazio alle nuove generazioni, selezionate positivamente secondo i canoni darwiniani. Anche le cellule e le molecole che compongono il nostro organismo invecchiano e muoiono. Per es., le proteine del sangue (anticorpi, ormoni, ecc.) vengono progressivamente modificate chimicamente finché non sono riconosciute da opportune cellule ‘spazzino’ e rimosse dal circolo; le estremità dei nostri cromosomi tendono ad accorciarsi a ogni divisione cellulare, sino a che questa viene compromessa; i mitocondri, le centrali di produzione energetica all’interno delle cellule, vanno incontro a una progressiva perdita di efficienza. Queste considerazioni ci aiutano a capire almeno in parte perché Dolly (la pecora clonata a partire da una cellula estratta da un animale adulto) morì giovane. In questo contesto di progressivo invecchiamento dei sistemi molecolari e cellulari si instaura la n., cioè la progressiva perdita di funzione o morte di neuroni o cellule accessorie, con conseguente compromissione dell’intero sistema nervoso, in primis il cervello. Essa si manifesta con modalità molto diverse dando luogo a patologie ben distinte tra loro dal punto di vista clinico, istologico e biochimico. Tra queste ricordiamo la malattia di Alzheimer, il morbo di Parkinson, la corea di Huntington, le atassie, la sclerosi laterale amiotrofica, le encefalopatie spongiformi, le demenze familiari, la demenza frontotemporale, la cosiddetta CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, arteriopatia cerebrale autosomica dominante con infarti sottocorticali e leucoencefalopatia) e molte altre. Ci sono molte forme di demenza legate alla n. che vengono comunemente riscontrate in individui anziani e che non sono associabili a queste o ad altre patologie fino a oggi descritte, in larga misura a causa del limitato livello di conoscenza.
Eziologia della neurodegenerazione
Nella maggior parte dei casi le malattie neurodegenerative sono sporadiche, ossia non dovute a mutazioni genetiche precise: quindi, parenti e figli di questi pazienti hanno la stessa probabilità di sviluppare la malattia della popolazione media. Pochi casi sono invece genetici, ossia hanno la loro causa in mutazioni ben precise, generalmente autosomiche dominanti, che si trasmettono con alta penetranza. Tali patologie sono caratterizzate da insorgenza precoce e coinvolgono molti familiari del paziente (il figlio di un soggetto affetto da malattia neurodegenerativa a trasmissione autosomica dominante ha il 50% di probabilità di sviluppare la stessa malattia). Nella malattia di Alzheimer solo l’1% dei casi diagnosticati è genetico, mentre nella malattia di Parkinson, nella SLA, nelle encefalopatie spongiformi e nella demenza frontotemporale i casi genetici sono il 5÷10%, 10%, 10÷20% e 20÷30%, rispettivamente. Alcune patologie, tra cui la corea di Huntington e la CADASIL, sono invece prevalentemente genetiche dominanti. Le cause delle forme sporadiche di malattie neurodegenerative sono essenzialmente ignote anche per le patologie sino a oggi più studiate. Tra i fattori di rischio che aumentano l’incidenza dell’Alzheimer, per es., ci sono il trauma cranico, il basso livello di educazione e di utilizzo delle funzioni cerebrali in età avanzata, l’invecchiamento precoce dei mitocondri, lo stress ossidativo, le infiammazioni croniche, il basso livello di estrogeni, l’ipercolesterolemia, l’ipertensione arteriosa, l’obesità e il diabete. Sono stati identificati anche fattori di rischio genetici, ossia la presenza di certe forme alleliche di alcuni geni che aumentano il rischio di sviluppare le malattie senza esserne la causa principale: il caso più conclamato è l’allele e4 del gene codificante l’apolipoproteina E. La nostra conoscenza dei fattori di rischio resta comunque assai limitata anche per una malattia molto studiata come l’Alzheimer. Molti fattori di rischio restano da identificare e non si conoscono bene neppure i meccanismi molecolari e cellulari con cui quelli già identificati aumentano il rischio di Alzheimer; ancor meno note sono le interconnessioni e le forme di cooperatività positiva o negativa tra questi fattori di rischio. La capacità di stimare per un individuo la probabilità di insorgenza di Alzheimer, l’età di comparsa della malattia e la severità della stessa restano pressoché nulle.
Malattie neurodegenerative trasmissibili
Sia nelle forme sporadiche sia in quelle genetiche, le malattie neurodegenerative non sono trasmissibili, a eccezione di alcune encefalopatie spongiformi; queste malattie sono, al pari di molte altre, prevalentemente sporadiche con una minoranza di casi genetici, ma possono essere trasmesse da altri individui umani o animali affetti dalla stessa patologia. Popolazioni tribali delle Nuova Guinea dedite fino a pochi anni fa alla pratica del cannibalismo rituale, che prevedeva di cibarsi del cervello del congiunto morto per permetterne la sopravvivenza dell’anima, erano affette da una forma nota col nome di kuru (➔). Analogamente l’encefalopatia spongiforme bovina (la cosiddetta sindrome della mucca pazza, ➔ encefalopatia spongiforme bovina) e, nell’uomo, la variante della malattia di Creutzfeldt-Jakob, insorgono in bovini ed esseri umani che si alimentano con alcune parti di bovini affetti.
Patogenesi delle malattie neurodegenerative e aggregati proteici
Un aspetto che riguarda molte patologie neurodegenerative e che può essere assurto a loro minimo comune denominatore, è l’aggregazione di una data proteina, caratterizzata da struttura aberrante (misfolded), a formare depositi che si accumulano nel cervello. Indipendentemente dalla proteina che li costituisce e dal compartimento in cui si accumulano, gli aggregati hanno spesso un aspetto molto simile, tipicamente fibrillare (struttura cross-β); in altri casi gli aggregati hanno invece una diversa natura strutturale e morfologica, spesso non del tutto identificata; esistono poi forme neurodegenerative nelle quali l’associazione tra aggregati e malattia rimane ancora da definire. Molte conoscenze sui meccanismi patogenetici alla base di queste malattie sono state acquisite dallo studio delle forme neurodegenerative genetiche o trasmissibili, sintomatologicamente simili alle più frequenti forme sporadiche corrispondenti. Spesso le mutazioni sono autosomiche dominanti e colpiscono geni che codificano proteine espresse nel sistema nervoso, favorendone la tendenza ad aggregare. È stata proprio l’identificazione di queste mutazioni ad associare l’aggregazione della proteina coinvolta con la causa della patologia corrispondente. In alcuni casi la mutazione non colpisce direttamente la proteina che aggrega, ma un’altra molecola che ne aumenta la produzione o la modifica favorendone l’aggregazione (come nella maggioranza delle mutazioni associate all’Alzheimer). Altre volte si hanno invece alterazioni delle funzioni cellulari preposte alla distruzione degli aggregati (come riscontrato per mutazioni associate alla malattia di Parkinson). L’indebolimento progressivo dei meccanismi cellulari protettivi è uno dei motivi per cui l’invecchiamento si correla all’aggregazione di proteine normalmente solubili. Questo problema è oggetto di intenso dibattito e studio, comunque si ritiene che l’aggregazione sia l’evento centrale nella patogenesi della malattia. Appare sempre più chiaro, inoltre, che non sono le fibrille mature a causare disfunzione cellulare, bensì gli aggregati solubili di piccola taglia (oligomeri) che si formano prima di quelli maturi e che possono essere da questi rilasciati. La trasmissione dominante di molti casi genetici suggerisce che gli aggregati – quale che sia la forma più nociva – acquisiscano una nuova funzione tossica all’interno dell’organismo, operando azione di ‘sabotaggio’ nei confronti delle normali funzioni cellulari (gain of toxic function). In questi casi, la natura nociva dell’aggregazione non risiede quindi nella sola sottrazione della forma normale e funzionalmente attiva della proteina in questione. La tossicità di aggregati proteici ricostituiti in vitro nei confronti di cellule in coltura e modelli animali suffraga questa ipotesi. I meccanismi con cui gli aggregati proteici causano disfunzionamento cellulare rimangono ancora oggetto di studio. È probabile che siano comunque non solo dipendenti dalla patologia in questione, ma anche diversi di cellula in cellula nella stessa patologia.
Ripiegamento (folding) delle proteine
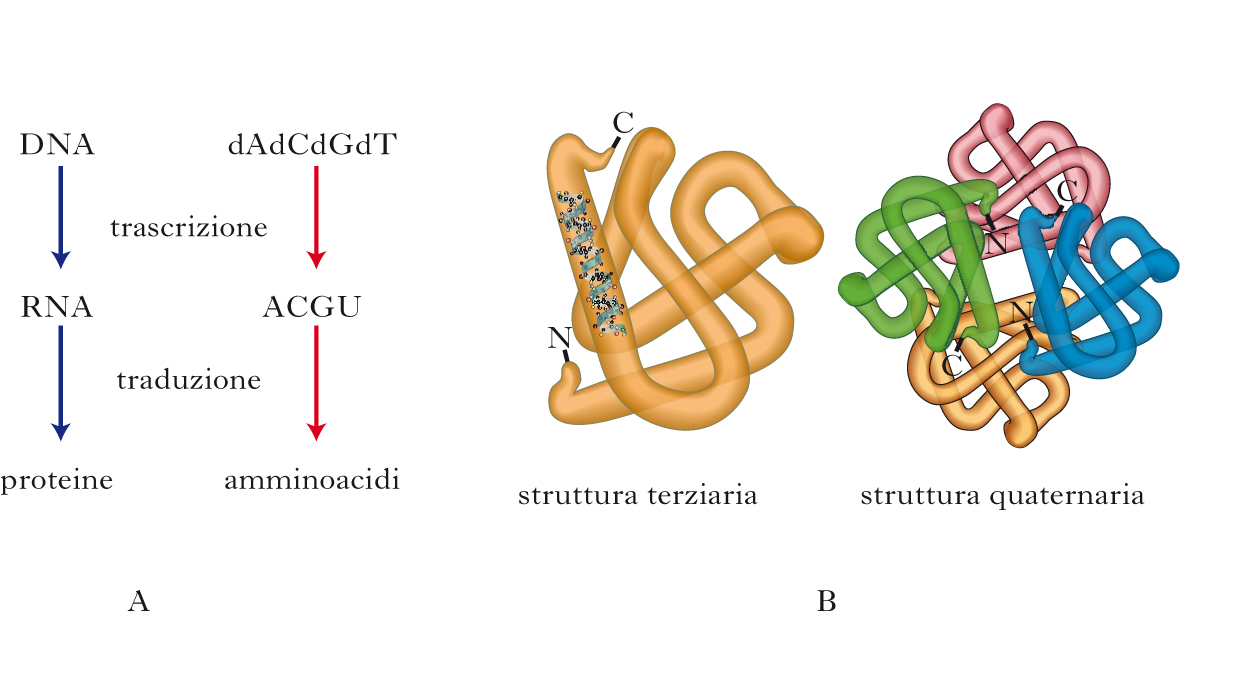
Il cosiddetto dogma centrale della biologia stabilisce che l’informazione genetica è contenuta e tramandata nel DNA, trascritta nell’RNA e infine tradotta in proteine, che costituiscono il principale hardware della materia vivente. DNA e RNA sono costituiti da sequenze lineari di nucleotidi, che formano un alfabeto composto di 4 sole lettere, ognuna corrispondente a un dato nucleotide. Le proteine o polipeptidi sono invece ‘collane’ di 20 diversi tipi di amminoacidi, la sequenza dei quali determina una certa funzione biologica. Nella seconda metà del secolo scorso, sono stati risolti la natura del codice genetico e i meccanismi mediante i quali l’informazione genetica viene tramandata di generazione in generazione (replicazione) e posta in opera nei singoli individui (trascrizione da DNA a RNA e traduzione da RNA a proteine). Un amminoacido è codificato da una sequenza di tre nucleotidi (tripletta). Dato che i nucleotidi sono di 4 tipi diversi, le triplette possibili sono 43, cioè 64. Considerando che gli amminoacidi sono solo 20, ne segue che alcuni amminoacidi sono codificati da più di una tripletta. Alcune triplette inoltre costituiscono un segnale di arresto al processo di traduzione. Grazie a questo codice genetico l’informazione contenuta nel DNA (ossia nei geni) viene codificata in proteine. Tuttavia, per svolgere la sua funzione biologica una certa proteina deve essere caratterizzata da una determinata struttura tridimensionale, e quindi è necessario che la sequenza amminoacidica che la compone si ripieghi nello spazio. Solo una data conformazione spaziale può svolgere la funzione biologica a cui è destinata. In accordo col dogma centrale, l’informazione necessaria al ripiegamento è presente nella sequenza primaria di amminoacidi. A tutt’oggi sappiamo ben poco sui meccanismi che consentono il ripiegamento (folding) di una proteina: si calcola che una proteina di 100 amminoacidi possa assumere 1030 possibili conformazioni tridimensionali, delle quali solo una è quella funzionale (nativa), che normalmente corrisponde a quella termodinamicamente più stabile. Se il ripiegamento procedesse per tentativi, alla rimarchevole velocità di 1013 tentativi al secondo, occorrerebbero 1017 secondi, e cioè l’età stimata dell’Universo, per ripiegare una proteina! Questo paradosso, noto come paradosso di Levinthal dal nome dello scienziato che lo ha formulato, illustra bene la centralità di questo problema biologico. È chiaro come debbano esistere meccanismi di folding, che molti definiscono il secondo codice genetico. Altrettanto chiaro è che un processo di tale complessità va incontro a errori, le cui conseguenze sono appunto molte patologie neurodegenerative e altre malattie ‘conformazionali’, nelle quali una specifica proteina assume forma aberrante.
Errori di folding e malattie neurodegenerative
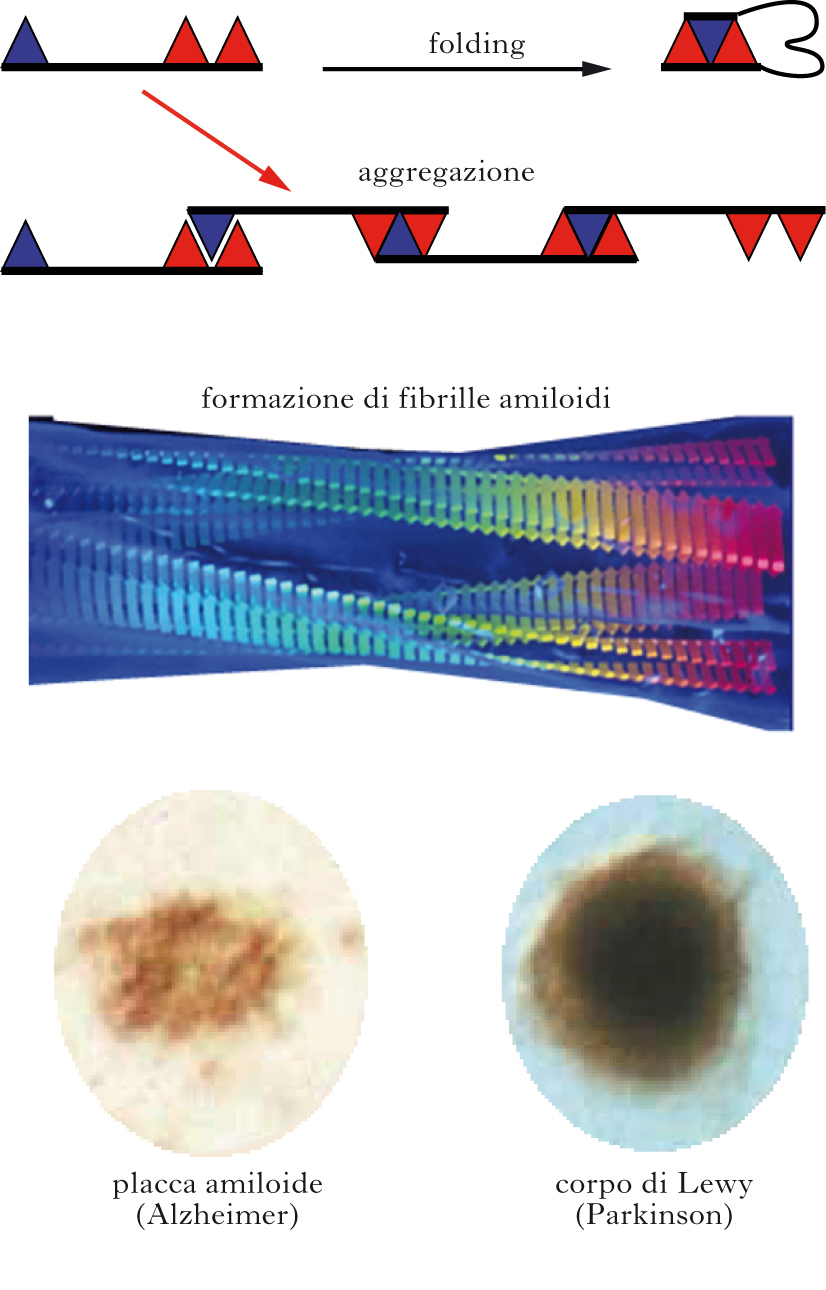
I meccanismi precisi con cui una proteina si ripiega sono ancora poco chiari. Schematizzando al massimo, il folding avviene giustapponendo porzioni che hanno affinità chimica l’una per l’altra formando legami deboli di varia natura. In tal modo, il processo di folding non procede solo per tentativi e avviene in tempi rapidi. In alcune condizioni però, quali per es. una concentrazione eccessiva di proteina o la presenza di mutazioni che destabilizzano lo stato nativo o aumentano la tendenza all’aggregazione, il legame non avviene all’interno della stessa molecola proteica (legame intramolecolare) ma tra zone affini di molecole proteiche distinte (legame intermolecolare). Si formano così dimeri, poi trimeri, quindi oligomeri più grandi fino ad arrivare ad aggregati complessi, ossia piccole fibrille. Le estremità delle fibrille agiscono da centri di nucleazione permettendo l’aggiunta di nuove molecole proteiche dello stesso tipo e consentendo quindi la crescita delle fibrille. Le fibrille possono anche rompersi generando nuove estremità. In altre parole l’aggregazione di nuove molecole proteiche è facilitata dalla presenza di fibrille preformate (fenomeno detto di seeding). In questo scenario si può comprendere la trasmissibilità della malattia di Creutzfeldt-Jacob. Ingerendo fibrille preformate, queste agiscono da stampo o seme (seed), catalizzando la formazione di nuove fibrille. Il meccanismo ricorda la nucleazione del ghiaccio: l’aggiunta di piccoli cristalli preformati (seme cristallino) a una soluzione di acqua fredda accelera enormemente il processo di gelificazione.
Terapia e diagnosi precoce
I margini d’intervento terapeutico nei confronti delle malattie neurodegenerative sono purtroppo assai ridotti e riguardano per lo più approcci in corso di sperimentazione. Per la sola malattia di Alzheimer si contano attualmente (2010) almeno dieci filoni di sperimentazione, che vanno da un sorta di vaccinazione attiva (nel tentativo di ottenere anticorpi che prevengono la formazione di aggregati tossici) all’uso di inibitori selettivi delle β- e γ-secretasi, le due proteasi che generano il peptide Aβ dalla proteina precursore amiloide, che va a formare aggregati e fibrille. Strade seguite sono la messa a punto di nuovi farmaci o chelanti ionici capaci di ritardare l’aggregazione proteica o l’utilizzo di neuroprotettori, antinfiammatori e antiossidanti in grado di favorire la sopravvivenza di sinapsi e neuroni. La speranza di intervento non è affidata alla sola terapia, ma anche alla diagnosi precoce. È stata evidenziata una serie di biomarcatori in grado di manifestarsi prima dello sviluppo della malattia di Alzheimer e che quindi potrebbero fornire indicatori per intervenire precocemente. Fabrizio Chiti, Roberto Sitia