
neurodegenerativo
neurodegenerativo
Che si riferisce a una degenerazione neuronale, cioè a un processo di distruzione, i cui prodotti inducono meccanismi di fagocitosi e di gliosi all’interno del tessuto nervoso stesso. I processi n. che portano alla morte dei neuroni possono originare da cause genetiche o da reazioni da invecchiamento, e corrispondono a molti quadri istopatologici (malattie n.). La genetica molecolare ha fatto progredire gli studi sulle malattie n., per spiegare il coinvolgimento selettivo di sistemi di neuroni correlati a ciascuna sindrome, e l’attivazione di geni cosiddetti della morte cellulare programmata. Anche il sistema della glia è coinvolto nelle malattie n., e molti studi si propongono di correlare il metabolismo neuronale, e le sue patologie, con l‘attività delle cellule gliali, la cui importanza si va sempre più affermando parallelamente alle acquisizioni sulla fisiopatologia dei neuroni.
Le malattie neurodegenerative
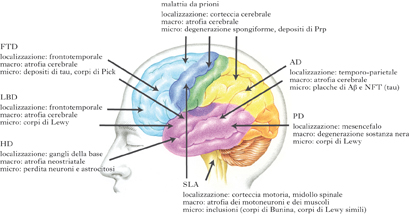
Le malattie neurodegenerative sono accomunate dalla perdita progressiva di popolazioni neuronali in specifici sistemi neuronali (non accompagnata da una franca infiammazione), da gliosi, da una patogenesi comune, da insorgenza subdola, dal progressivo e inesorabile decadimento delle condizioni fisiche e mentali. In base alla localizzazione delle lesioni nel sistema nervoso, è possibile distinguere forme che interessano la corteccia cerebrale, ossia morbo di Alzheimer (AD), morbo di Pick, morbo di Creutzfeldt-Jacob, demenza frontotemporale (FTD), demenza con corpi di Lewy (LBD); forme che interessano i gangli della base, ossia morbo di Parkinson (PD), e morbo di Huntington (HD); forme che colpiscono i neuroni motori, ossia sclerosi laterale amiotrofica (SLA); e forme che colpiscono il midollo spinale e il cervelletto, queste ultime denominate genericamente degenerazioni spinocerebellari (malattia di Friedreich).
Sintomatologia
Nelle malattie neurodegenerative la morte dei neuroni avviene gradualmente nel corso di mesi o anche anni. In tale arco di tempo i neuroni rimasti, almeno in parte, sostituiscono nelle proprie funzioni quelli distrutti. I primi segni della malattia compaiono quando la perdita progressiva dei neuroni supera la capacità di compenso dei neuroni superstiti. I sintomi clinici maggiori che compaiono precocemente nelle forme a trasmissione genetica e più tardivamente nelle forme sporadiche sono: la demenza, cioè l’indebolimento progressivo e irreversibile di tutte le funzioni intellettive (memoria, attenzione, giudizio), e disturbi della condotta sociale (AD, morbo di Pick, LBD, FTD, morbo di Creutzfeldt-Jacob, ecc.); i disturbi del movimento, di tipo ipocinetico (PD e SLA) e di tipo ipercinetico (HD); atassia (eredodegerazioni spinocerebellari, atrofie cerebellari degenerative).
Eziologia
Solo una piccola percentuale delle malattie neurodegenerative è dovuta a mutazioni geniche che seguono una trasmissione ereditaria di tipo mendeliano. In genere la mutazione genica si associa alla presenza di una proteina che perde le sue funzioni originarie o acquista nuove funzioni. In un caso o nell’altro queste si rivelano fatali per la cellula colpita. Nelle forme sporadiche la causa è invece sconosciuta; diversi studi propendono per un’origine multifattoriale, in cui interagiscono fattori ambientali e predisposizione genetica. Il rischio di insorgenza di PD aumenta nei soggetti sottoposti a esposizione cronica di pesticidi, erbicidi, insetticidi, ai metalli pesanti o agli idrocarburi e solventi (dopo i 50 anni sarebbero ad alto rischio meccanici, verniciatori e restauratori). Anche alcune neurotossine come la MPTP (1-metil-4-fenil-1,2,3-tetradidropiridina, un composto che si forma durante la sintesi dell’eroina sintetica) e l’annonacina (un composto contenuto nel frutto guanabana largamente utilizzato dagli abitanti delle Guadalupe) possono causare sindromi simil-parkinsoniane. L’alta incidenza della SLA nell’isola di Guam e tra gli sportivi ha fatto ipotizzare che, anche per questa malattia, le abitudini alimentari (uso di farine ricche di amminoacidi in grado di attivare i recettori del glutammato) o il tipo di allenamento e l’abuso di integratori alimentari negli sportivi possano rappresentare forti fattori di rischio. Per quanto riguarda l’Alzheimer sporadico sono state considerate l’origine virale, l’intossicazione da alluminio, l’ipotesi di un aumento della permeabilità della barriera ematoencefalica alle sostanze tossiche e l’origine multifattoriale.
Patogenesi
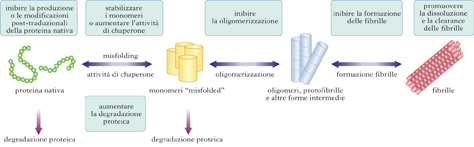
Malattie neurodegenerative come AD, PD o SLA condividono un meccanismo patogenetico comune che implica l’aggregazione e il deposito in sede extracellulare o intracellulare di aggregati proteici filamentosi e insolubili con caratteristiche ultrastrutturali simili alla sostanza amiloide. L’amiloidosi ha origine da una proteina nativa solubile che, a causa di un ripiegamento strutturale sbagliato (misfolded), che modifica la sua conformazione, si associa con sé stessa per formare aggregati di complessità crescente (oligomeri, protofibrille e fibrille). Gli aggregati possono essere costituiti dal peptide Aβ (placche senili nell’AD), dalla proteina tau (grovigli neurofibrillari nell’AD, nella FTD, nell’LBD, ecc.), dalla α-sinucleina (nel PD). Una data patologia può condividere più tipi di aggregati, come la sindrome di Down e la SLA/demenza/Parkinson di Guam, considerate triple amiloidosi perché presentano depositi di Aβ, tau e α-sinucleina. Una doppia amiloidoisi (Aβ, tau) è invece comune nella maggior parte dei pazienti di AD e in alcuni casi di PD (tau e α-sinucleina). Diversi fattori possono influenzare il bilancio tra proteina nativa, proteina misfolded, oligomeri e fibrille: l’aumentata produzione di proteine amiloidogeniche; non appropriate modificazioni post-traduzionali (in termini di fosforilazione, glicosilazione, digestione proteolitica); aumentati livelli di stress ossidativo per alterato metabolismo energetico (inquinanti ambientali e neurotossine come la MPTP danneggiano la catena respiratoria dei mitocondri) e per riduzione delle proteine antiossidanti; inefficacia dei sistemi proteolitici preposti alla rimozione di proteine danneggiate (sistema ubiquitina-proteosoma); insufficiente attività delle proteine chaperonine (proteine che facilitano la corretta organizzazione tridimensionale di altre proteine, il loro inserimento in una membrana e la loro organizzazione in strutture più complesse). I meccanismi attraverso cui gli aggregati proteici causano disfunzione e morte neuronale non sono ancora noti. Diversi dati suggeriscono che essi possono danneggiare il trasporto assonale, inibire il sistema ubiquitina-proteosoma, compromettere la trascrizione del DNA, aumentare i livelli di stress ossidativo e causare apoptosi. La prospettiva di un meccanismo patogenetico comune che raggruppa le malattie neurodegenerative nelle amiloidosi cerebrali apre possibilità di nuovi approcci terapeutici applicabili a un ampio spettro di malattie neurodegenerative.