
medulloblastoma
medulloblastoma
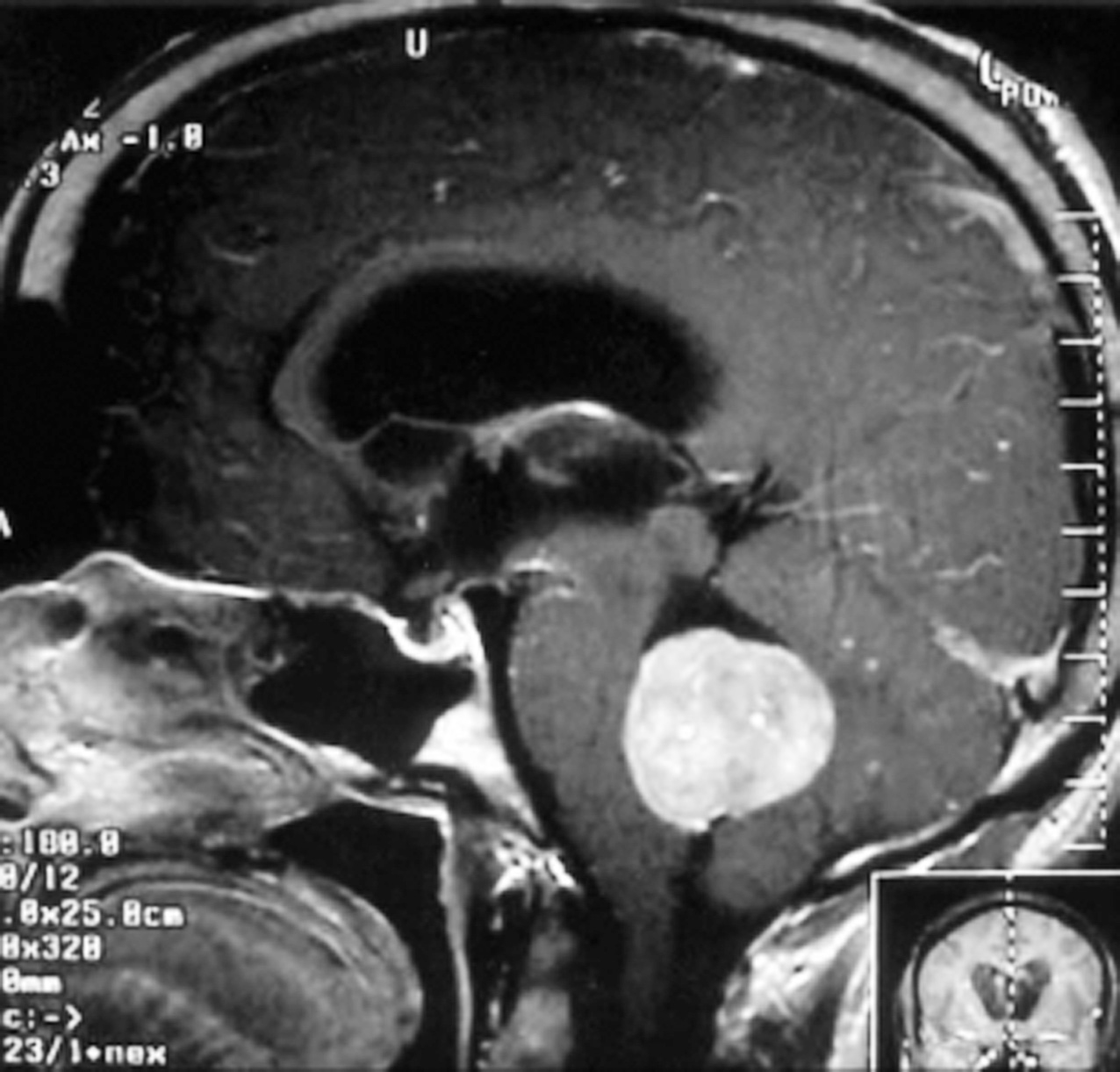
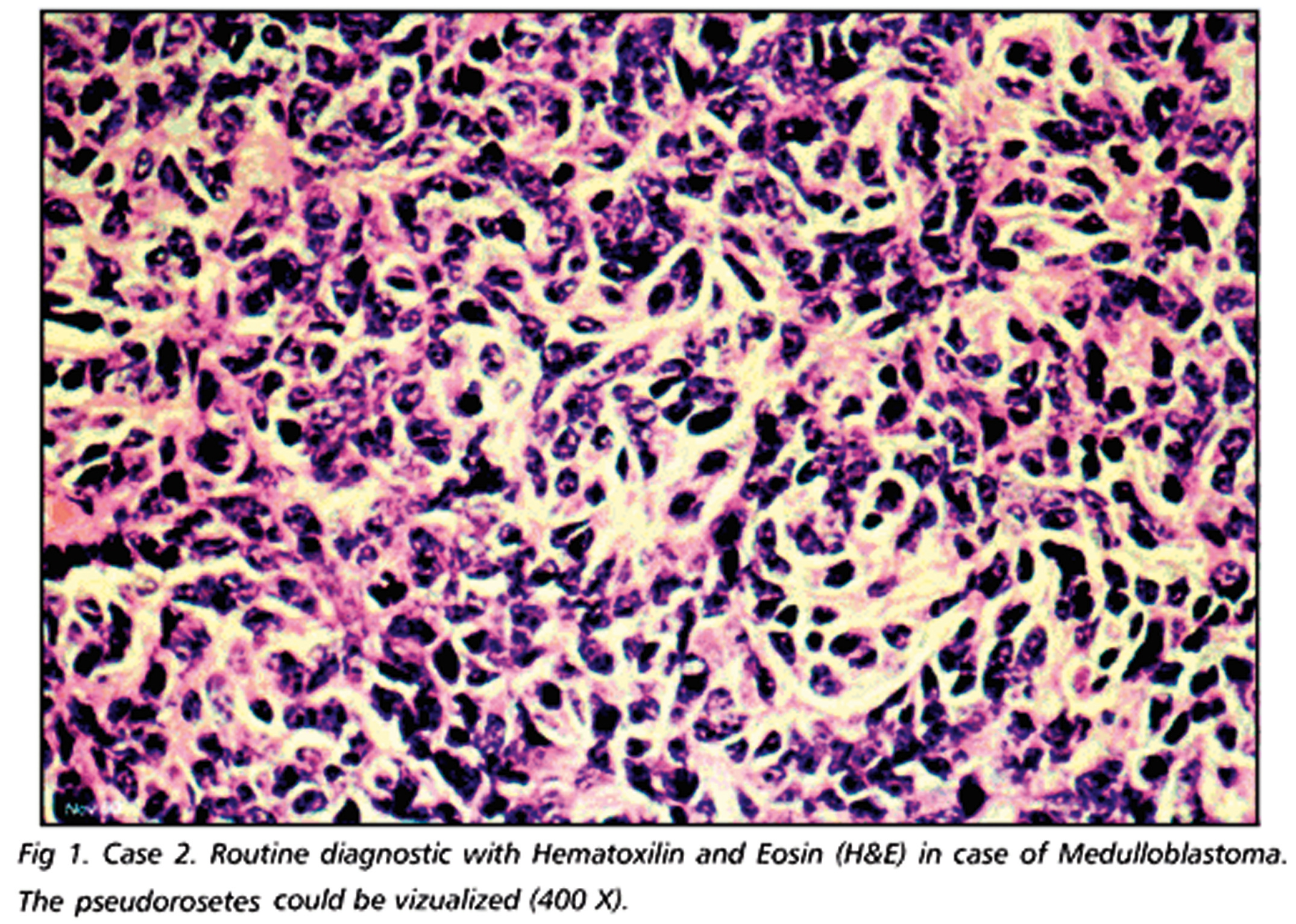
Il più comune tumore cerebrale maligno dell’infanzia; per definizione insorge nel cervelletto, a livello del verme, e meno frequentemente interessa gli emisferi cerebellari. La sintomatologia più frequente è legata allo sviluppo di atassia e di idrocefalo (➔) – presente in ca. il 70% dei casi – o di ipertensione endocranica, nonché alle frequenti metastasi midollari a livello toracico e lombare. La valutazione dell’estensione della neoplasia si effettua per mezzo di RMN con gadolinio (enhancement intenso, spesso disomogeneo). Il m. è costituto istologicamente da cellule indifferenziate piccole, ipercromiche, con scarso citoplasma, che talvolta circondano aree fibrillari acellulari per formare le cosiddette rosette neuroblastiche di Homer- Wright. La neoplasia mostra in genere aspetti differenziativi neuronali, meno frequentemente di tipo gliale.
Tipi di medulloblastoma
Esiste una variante, chiamata m. desmoplastico, caratterizzata da aree ricche in reticolo e collagene che delimitano noduli costituiti da cellule con maggiore differenziazione neuronale. Un’altra variante è il m. a grandi cellule (o anaplastico), costituito da cellule voluminose con nucleo vescicoloso e nucleoli prominenti e con marcate atipie e gigantismi nucleari.
Caratteristiche ed eziologia
Il m. infiltra diffusamente il parenchima cerebellare e lo spazio subaracnoideo. Esso tende a disseminare cellule per via liquorale dando metastasi nel sistema ventricolare e, a distanza, metastasi intradurali a livello del midollo spinale. Diverse modificazioni genetiche ed epigenetiche sono state descritte nel medulloblastoma. L’alterazione cromosomica più comune è la perdita del braccio corto del cromosoma 17 causata dalla duplicazione del braccio lungo del cromosoma 17 (isocromosoma 17q). Un’altra alterazione genetica è l’amplificazione dell’oncogene MYC. Ambedue queste alterazioni genetiche, quando presenti, indicano un decorso clinico più aggressivo. Le alterazioni molecolari alla base del m. sono quelle che interessano la morfogenesi del cervelletto. Si ritiene che la maggior parte dei m. origini dalla trasformazione neoplastica di precursori delle cellule dei granuli cerebellari (PGC), sebbene alcune di queste neoplasie possano anche originare da precursori di cellule nervose della regione subventricolare. Una delle principali vie di trasduzione del segnale deregolato è rappresentata da SHH (Sonic HedgeHog). Mutazioni germinali in componenti di SHH (Ptc e il repressore di Gli-SUFU) sono responsabili della sindrome di Gorlin, caratterizzata da una aumentata suscettibilità a sviluppare medulloblastomi. Mutazioni somatiche dei geni Ptc, Sufu e Smo sono osservate nel 25% dei casi sporadici di m., così come attivazioni epigenetiche dell’espressione del gene Gli-1. La via di trasduzione di SHH rappresenta uno dei principali regolatori di PGC, dal momento che SHH, un ligando agonista secreto dalle cellule di Purkinje, promuove la proliferazione dei progenitori delle cellule dei granuli nello strato granulare esterno (EGL) a seguito del legame al recettore Ptch; ciò suggerisce che l’attivazione incontrollata di questo segnale mitogenico sostenga l’oncogenesi. La via di trasduzione del segnale di WNT, in modo simile a SHH, è coinvolta in diversi processi dello sviluppo e dell’oncogenesi; mutazioni delle proteine WNT sono state descritte in circa il 15% dei m. sporadici.
Terapia
I trattamenti attuali includono resezione chirurgica totale, radioterapia dell’intero nevrasse e chemioterapia. La sopravvivenza a cinque anni con gli attuali trattamenti è ca. del 40% in bambini con m. metastatico o non operabile, e del 75% in pazienti con patologia di medio rischio. In aggiunta, la morbidità correlata al trattamento può essere severa; i bambini trattati possono presentare gravi deficit cognitivi e fisici a lungo termine.