
Materia soffice
Materia soffice
La locuzione materia soffice viene generalmente utilizzata per definire i materiali che, pur presentandosi in fase condensata, non sono né liquidi semplici né solidi cristallini. La m. s. occupa infatti una posizione intermedia tra lo stato liquido, nel quale le molecole sono in grado di scambiare liberamente le loro posizioni, e lo stato solido cristallino, nel quale sono disposte ai vertici di un reticolo. Nei materiali soffici le molecole, pur senza occupare posizioni fisse, sono in qualche modo vincolate e non hanno la libertà di scambiarsi tra loro. La m. s. riveste una grande importanza nella vita quotidiana: infatti è presente, per es., nelle pitture, negli adesivi e nei saponi e, anche, in gran parte degli alimenti. E il suo ruolo risulta cruciale nel settore delle scienze della vita. Altri materiali soffici si ritrovano in importanti processi industriali, come i polimeri fusi che vengono estrusi per formare oggetti di plastica.
La prima grande classe di materiali soffici comprende i polimeri costituiti da macromolecole in cui un numero elevato di unità di tipo idrocarburico sono unite tra loro per formare catene flessibili. Una seconda grande classe è rappresentata dai colloidi, dove una fase finemente suddivisa in particelle che possono avere dimensioni comprese tra qualche decina di nanometri e qualche micron è dispersa in una fase continua. Esistono diversi tipi di tali dispersioni, a seconda della forma fisica della fase dispersa: se essa è solida la dispersione si chiama sol; se invece è costituita da goccioline di un liquido insolubile nel liquido che compone la fase continua si è in presenza di un'emulsione; infine, quando la fase dispersa è costituita da bollicine di gas la dispersione viene denominata schiuma. Un'altra importante categoria è rappresentata dalle soluzioni di tensioattivi, che sono solitamente composti da molecole anfifiliche, contenenti una porzione fortemente idrofilica, che tende a sciogliersi nell'acqua, e un'altra marcatamente idrofobica, cioè respinta dall'acqua. In soluzione acquosa le parti idrofobiche di diverse molecole di tensioattivo si aggregano tra loro per generare particolari strutture dette micelle. Molto importanti dal punto di vista scientifico e applicativo sono anche i cristalli liquidi, in cui strutture molecolari anisotrope possono generare stati caratterizzati da ordinamenti intermedi tra quelli di un liquido semplice e di un solido cristallino.
Il termine soffice si riferisce alle proprietà meccaniche macroscopiche di questo tipo di materiali, che sotto determinate condizioni sono in grado di fluire. In generale, essi presentano proprietà viscoelastiche, manifestando cioè sia la tendenza a fluire, peraltro propria dei fluidi viscosi, sia la capacità di deformarsi se sottoposti a sollecitazione, tipica dei fluidi elastici. Sono quindi nettissime le differenze rispetto al comportamento di un solido cristallino, le cui molecole appaiono strettamente impacchettate. Se un solido cristallino viene sottoposto a compressione, le sue molecole vengono spinte le une verso le altre provocando un'eccitazione atomica che richiede un'energia nell'ordine di qualche elettronvolt ad atomo. Compressioni anche minime sono quindi associate a energie molto vicine a quelle di legame tra gli atomi e di conseguenza i solidi sono soggetti a frattura fragile. I materiali soffici, al contrario, sono in grado di sopportare compressioni molto elevate senza subire alcun danno visibile, nonostante il fatto che la loro energia di frattura sia molto minore. Si consideri, per es., un gel polimerico, solitamente caratterizzato da un'elevata resilienza, ossia da un'elevata capacità di resistere agli urti che può essere correlata alla scarsa connettività a livello molecolare del materiale. Le molecole di polimero formano un reticolo che può essere facilmente distorto nell'istante in cui il gel viene compresso. Le singole macromolecole sono flessibili, e i legami molecolari al loro interno possono ruotare e quindi consentire un allungamento delle catene senza che si verifichino le distorsioni atomiche tipiche dei solidi cristallini. I gel polimerici inoltre hanno densità piuttosto basse e questo consente alle catene polimeriche di disporre di uno spazio sufficiente per potersi distorcere liberamente senza provocare addensamenti molecolari locali. Si deve anche tenere conto del fatto che ogni catena polimerica non è fissata in un'unica configurazione, ma fluttua da una configurazione casuale a un'altra. Poiché i legami nella catena hanno orientamenti indipendenti, per certe dimensioni caratteristiche il cammino descritto da una catena polimerica nello spazio è casuale. Una compressione ha l'effetto di ridurre queste dimensioni limitando il numero di configurazioni possibili. La distorsione del polimero, così, provoca una diminuzione di entropia, la quale richiede che venga eseguito del lavoro sul sistema. Nella molecola di polimero inoltre si produce una tensione. In ogni caso le energie in gioco sono di gran lunga minori di quelle richieste nelle compressioni dei solidi cristallini, e sono nell'ordine di kT (k è la costante di Boltzmann e T la temperatura assoluta), che rappresenta l'energia termica e che, a temperatura ambiente, vale all'incirca 1/40 di elettronvolt.
Proprietà
I vari materiali soffici, pur così diversi tra loro, hanno in comune alcune caratteristiche molto significative. Anzitutto assumono una grande importanza le scale di lunghezza che definiscono le loro strutture, comprese tra le dimensioni atomiche e quelle macroscopiche, e che vengono chiamate mesoscopiche. Le particelle colloidali sono tipicamente più piccole di un micron, le catene polimeriche hanno lunghezze nell'ordine di alcune decine di nanometri. I modelli teorici utilizzati per descrivere questi materiali devono tenere conto di questa varietà di dimensioni, emancipandosi dai dettagli relativi alla scala atomica. È interessante notare che emerge un comportamento universale comune a varie sostanze: per es., molti aspetti del comportamento dei polimeri non derivano dalle caratteristiche chimiche delle singole unità che li compongono, ma da quelle topologiche derivanti dal fatto che una molecola di polimero è lunga, flessibile e non può essere attraversata da altre catene. Un tipico esempio è rappresentato dalla viscosità di un polimero, che dipende, secondo una legge di potenza, dal peso molecolare delle catene. Anche le proprietà ottiche di una dispersione dipendono dalle dimensioni medie delle particelle che la compongono secondo una legge di potenza. Anche le fluttuazioni termiche inoltre sono un fattore molto importante nel determinare le caratteristiche dei materiali soffici. Infatti, pur essendo più grandi delle dimensioni atomiche, le unità strutturali che compongono i materiali soffici sono in genere sufficientemente piccole da subire gli effetti del moto browniano. Non solo, ma anche le tipiche energie associate con la formazione delle strutture presenti in un materiale soffice sono confrontabili, in ordine di grandezza, all'energia termica. Essi dovrebbero essere, quindi, visualizzati come soggetti a un moto continuo: le molecole di polimero in soluzione si contorcono e girano in continuazione, mentre le membrane formate da strati di molecole anfifiliche non sono rigide ma si muovono e si piegano sotto l'effetto del moto browniano.
Infine, i materiali sono in generale caratterizzati dalla capacità di formare strutture a livello mesoscopico. Grazie all'importanza rivestita dal moto browniano i materiali soffici sono in grado di evolvere verso l'equilibrio, che tuttavia deve essere concepito in maniera dinamica. Inoltre i sottili bilanci tra fattori entalpici ed entropici spesso generano un comportamento di fase molto variegato, in cui possono formarsi spontaneamente strutture anche molto complesse. A un livello superiore spesso si formano strutture sopramolecolari.
Dinamica
I tipi di strutture che si possono presentare nei materiali soffici sono molto diverse. Esistono rimarcabili fenomeni di organizzazione intramolecolari. Un esempio significativo è rappresentato dalla struttura a elica assunta da molti polipeptidi, dovuta alla formazione di legami idrogeno tra unità monometriche vicinali, con un meccanismo non dissimile da quello che governa la formazione della doppia elica del DNA.
Tra i fenomeni di riordinamento più importanti offerti da sistemi costituiti da molecole di un unico tipo ci sono quelli che si possono presentare nei copolimeri fusi. Un copolimero è una catena polimerica che non è chimicamente omogenea, ma presenta unità monometriche di due tipi diversi, A e B. La successione dei due tipi di unità nella catena può essere alternata, quando a un'unità A fa sempre seguito una catena B, oppure casuale. Nei copolimeri a blocchi, le catene presentano due blocchi separati, uno composto interamente da unità di tipo A e l'altro da unità di tipo B. Se le due porzioni di un copolimero a blocchi non sono mutuamente solubili, allo stato fuso esse tendono a separarsi, ma ciò è impedito dal fatto che le due parti appartengono alla medesima catena. Di conseguenza nella fusione si creano dei microdomini, ognuno dei quali contiene la porzione A o la porzione B delle diverse catene copolimeriche. Aumentando le dimensioni dei domini diminuisce l'area interfacciale tra le regioni occupate dalle porzioni A e quelle occupate dalle porzioni B, con conseguente diminuzione di energia libera. Nel contempo, però, ciò provoca un allungamento delle catene riducendone l'entropia. Alla fine, equilibrando questi due contributi contrastanti, si ottengono strutture periodiche anche molto complesse, che dipendono dalle lunghezze relative delle porzioni A e B, ovvero dalla frazione volumetrica ϕA occupata dai blocchi A. Se ϕA è piccola, minore di 0,2, si formano dei domini sferici di A in una matrice di B, mentre se ϕA è circa pari a 0,3, la porzione A forma dei domini cilindrici. Quando invece le frazioni volumetriche occupate da A e B sono confrontabili, si generano strutture lamellari.
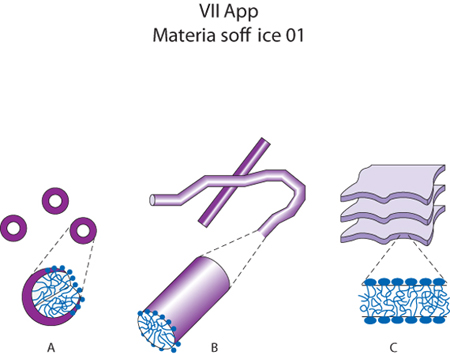
Come menzionato precedentemente, altri esempi piuttosto noti di organizzazione sono rappresentati dalla formazione di micelle nelle soluzioni acquose di tensioattivi. Questo processo è provocato dal cosiddetto effetto idrofobico, che giustifica perché acqua e sostanze idrocarburiche non siano mutuamente miscibili. Ciò deriva da un riordinamento locale delle molecole d'acqua in strutture tetraedriche che si crea ogni volta che molecole idrocarburiche vengono a trovarsi in un ambiente acquoso dando luogo a una diminuzione di entropia. La separazione di fase tra acqua e idrocarburi avviene solitamente su scala macroscopica, ma nel caso delle sostanze tensioattive, che presentano carattere anfifilico, tale effetto idrofobico provoca a livello mesoscopico la formazione di strutture, le micelle, che generalmente hanno dimensioni che non superano pochi nanometri. È la presenza di gruppi dotati di affinità marcatamente differenti nei confronti dell'acqua che dota i tensioattivi di proprietà anfifiliche. Spesso si tratta di una testa ionica attaccata su una catena idrocarburica. I tensioattivi più comuni sono dei sali di acidi grassi. Queste molecole, messe in soluzione acquosa, al di sopra di una certa concentrazione critica hanno propensione ad assemblarsi tra loro per formare strutture che solitamente sono sferiche, ma che possono assumere anche forme cilindriche o a bistrato (fig. 1). Esistono criteri geometrici piuttosto semplici per prevedere il tipo di struttura formata dalle molecole di tensioattivo, a seconda del valore assunto dal rapporto v/lca0, dove v rappresenta il volume occupato dalla coda idrocarburica, a0 l'area del gruppo di testa in corrispondenza della superficie dell'aggregato e lc la lunghezza critica della catena, correlata alla lunghezza della catena idrocarburica, completamente estesa.
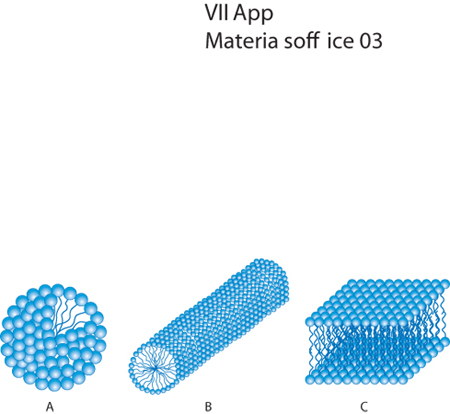
A differenza delle mesofasi liquido-cristalline quelle liotropiche si formano in soluzione, e la loro formazione è controllata dalla concentrazione di una specie che, solitamente, ha carattere anfifilico. Dei tensioattivi si è enfatizzata la capacità di formare micelle, ma in realtà essi presentano, al crescere della loro concentrazione, un comportamento di fase abbastanza variegato. Alle basse concentrazioni le micelle non mostrano alcun tipo di ordine legato alle loro posizioni, ma a concentrazioni un po' più elevate esse tendono a manifestare interazioni repulsive, che possono derivare da effetti sterici e di volume escluso. Di conseguenza le micelle cominciano a occupare lo spazio in maniera più efficiente, sistemandosi, per es., in corrispondenza di vertici di cubi, i cui lati hanno lunghezze nell'ordine di qualche decina di nanometri (fig. 2 A), quindi notevolmente maggiori delle dimensioni atomiche. L'effetto, per quanto concerne la forma fisica macroscopica e il comportamento dei materiali, è notevole, dato che in queste condizioni le soluzioni di tensioattivi assumono l'aspetto di un gel, con viscosità molto elevate, ed esibiscono un certo limite di scorrimento, per cui è necessario sottoporre il materiale a una sollecitazione minima affinché questo cominci a fluire. Aumentando ulteriormente la concentrazione del tensioattivo, le micelle assumono forma cilindrica e si organizzano su strutture esagonali, formando quindi cristalli liquidi di tipo colonnare (fig. 2 B). A concentrazioni ancora più alte le micelle formano dei bistrati piani, che costituiscono una fase smettica (fig. 2 C). In realtà la descrizione riportata è piuttosto semplificata, perché il comportamento di fase dei tensioattivi è molto complicato e dipende dalle dimensioni della testa idrofila e della coda idrofoba, dalla forza ionica della soluzione, dall'eventuale presenza di cotensioattivi eccetera.
Dispersioni colloidali e polimeri
Le due classi principali di materiali soffici, le dispersioni colloidali e i polimeri, possono essere considerate come modi attraverso i quali è possibile ottenere degli aggregati poliatomici. Nel primo caso, la forma dispersa è spesso sfruttata per ottenere materiali dotati di particolari caratteristiche fisiche. Per es., il nerofumo disperso nell'inchiostro, per la sua natura colloidale, manifesta un'efficiente assorbimento della luce incidente, oppure le dispersioni in cui granuli di magnetite vengono incapsulati in particelle di polistirene, a loro volta disperse in acqua, permettono di ottenere ferrofluidi ricoperti da polimero. Talvolta però la forma dispersa serve per impartire particolari proprietà chimiche ai materiali, come nel caso dei granuli fotosensibili nei film fotografici, o nei casi in cui le particelle disperse consentono di produrre meccanismi di riconoscimento molecolare.
Le dispersioni colloidali possono essere prodotte sminuzzando masse, più o meno grosse, di materiali mediante metodi meccanici. Le particelle ottenute in questo modo vengono successivamente disperse in una fase continua. Tuttavia non è semplice fornire sollecitazioni meccaniche sufficienti per ottenere particelle di dimensioni colloidali, specialmente quando la massa si trova allo stato solido. Inoltre le dispersioni ottenute in questo modo solitamente sono costituite da particelle dotate di un'ampia distribuzione granulometrica, e talvolta di forme irregolari. Pertanto in generale si preferisce produrre le dispersioni colloidali facendo ricorso a metodi in virtù dei quali, in qualche modo, le molecole di una sostanza, disciolte in una fase continua, vengono indotte a unirsi per formare la dispersione, come, per es., attraverso un processo di nucleazione. Operando in modo tale che la nucleazione si completi in tempi ristretti, è possibile ottenere dispersioni nelle quali le dimensioni delle particelle sono caratterizzate da una distribuzione delle dimensioni piuttosto stretta.
In ogni caso le particelle colloidali hanno la tendenza ad aggregarsi tra loro, sotto l'effetto delle forze attrattive di van der Waals le quali diventano sempre più consistenti a mano a mano che le particelle crescono di dimensioni. Lo stato disperso è, nella maggior parte dei casi, termodinamicamente sfavorito, poiché comporta, rispetto a quello massivo, una spesa energetica legata alla formazione della superficie interfacciale. Tuttavia è possibile stabilizzare la dispersione, creando una barriera di potenziale tra le particelle che deve essere almeno maggiore di kT, ovvero dell'energia associata ai moti molecolari. Essa può essere ottenuta mediante effetti di tipo elettrostatico, o di tipo sterico, oppure tramite la combinazione dei due, mediante effetti detti elettrosterici. La stabilizzazione elettrostatica solitamente viene ottenuta attaccando alla superficie delle particelle, mediante adsorbimento chimico o fisico, dei gruppi dotati di carica elettrica. In questo modo si creano cariche localmente non bilanciate che provocano l'insorgere di repulsioni coulombiane tra le particelle.
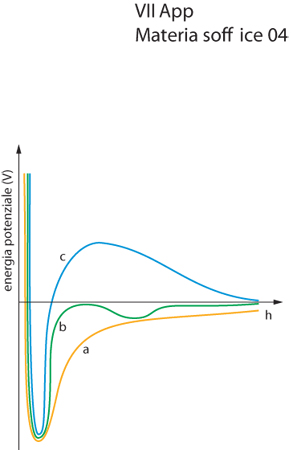
La situazione in realtà è un po' più complessa se le particelle sono disperse in acqua, poiché in acqua sono sempre presenti ioni, che schermano le interazioni elettrostatiche. Una particella carica in superficie attrae dei controioni, una parte dei quali si lega strettamente alla superficie della particella stessa formando lo strato di Stern, mentre la parte rimanente forma un profilo diffuso di concentrazione, creando un potenziale elettrostatico che, sotto determinate condizioni, può essere espresso con una funzione che decade esponenzialmente con il graduale allontanamento dalla superficie della particella. Combinando il potenziale attrattivo che deriva dalle forze di van der Waals con il potenziale repulsivo elettrostatico, è possibile ottenere il potenziale di interazione tra due particelle (fig. 3), secondo quanto stabilito dalla teoria DLVO (dalle iniziali di B. Derjaguin, L.D. Landau, E.J. Verwej e T. Overbeek). Poiché la rapidità con cui il potenziale elettrostatico decade all'allontanarsi dalla superficie della particella aumenta al crescere della forza ionica della fase continua, in una dispersione stabilizzata elettrostaticamente è possibile far prevalere le forze attrattive e indurre la flocculazione tra le particelle, aggiungendo quantità sufficienti di sali.
La stabilizzazione sterica delle dispersioni viene invece ottenuta facendo adsorbire delle molecole di polimero sulla superficie delle particelle. Avvicinando le particelle tra di loro si viene a provocare una compressione delle catene di questi polimeri, compressione che induce un meccanismo di repulsione osmotica: restringendo il numero di conformazioni accessibili a ogni catena, si provoca un aumento di entropia. Anche in questo modo è quindi possibile creare una barriera di potenziale alla flocculazione, che contrariamente alla stabilizzazione elettrostatica non è sensibile alla concentrazione di sali in soluzione, ma dipende dalle interazioni tra polimero e fase continua, e dalla temperatura. Generalmente i copolimeri a blocchi sono i sistemi più efficienti nell'impartire stabilizzazione sterica: un blocco deve avere affinità per la fase dispersa, l'altro per la fase continua. La stabilizzazione elettrosterica si ottiene mediante sostanze capaci di combinare i due meccanismi elettrostatico e sterico, e viene tipicamente ottenuta mediante polielettroliti.
Se si aggiunge una certa quantità di elettrolita a una dispersione stabilizzata elettrostaticamente, oppure se si varia la temperatura a una dispersione che è stabilizzata stericamente, è possibile indurre una destabilizzazione, provocando la flocculazione delle particelle. Le particelle, dotate di una certa energia termica, si spostano sotto l'effetto dei loro moti browniani e collidono, e poiché esse non sono più dotate di alcuna barriera che le stabilizzi, ogni collisione provoca flocculazione. Dapprima si formano doppietti e tripletti, poi clusters di dimensioni sempre maggiori che, a un certo punto, essendo troppo grandi per poter essere soggetti a moto browniano, sedimentano.
Un processo di flocculazione che ha attirato particolare attenzione e che è stato ampiamente studiato è il cosiddetto processo di crescita in regime diffusivo, in cui il cluster viene costruito aggiungendo fra loro le particelle a una a una, in condizioni di diluizione infinita. Si suppone che la flocculazione avvenga con una velocità molto elevata, al limite infinita, e di conseguenza la formazione del cluster è cineticamente limitata dalla diffusione delle particelle. In questi clusters le particelle si succedono seguendo un cammino casuale, formando dunque strutture altamente ramificate, aperte, apparentemente disordinate, tipiche dei sistemi frattali. Per questi tipi di flocculi è valida una legge di scala del tipo ak/a=kexp(1/Df), dove a è il raggio della singola particella, ak il raggio del flocculo, k il numero di particelle in esso contenuto e Df un parametro chiamato dimensione frattale, che vale 1,7 se tale aggregazione si realizza in uno spazio bidimensionale, e 2,5 se invece essa ha luogo in uno spazio tridimensionale. L'aggregazione in regime diffusivo ha molte affinità con svariati processi di crescita, seppure molto diversi dal punto di vista fisico, come la crescita di colonie batteriche.
La sintesi dei polimeri può essere concepita come un altro modo di formare strutture poliatomiche, legando tra loro molecole piccole in una catena flessibile. Le direzioni relative dei successivi legami tra le molecole sono piuttosto casuali. La correlazione direzionale tra i legami diventa pressoché trascurabile dopo poche lunghezze di legame, cosicché le proprietà statistiche di una lunga catena polimerica sono quelle di un cammino casuale. Tuttavia bisogna notare che il modello 'a cammino casuale' non descrive in modo completamente corretto la conformazione di un polimero in soluzione, e ciò è dovuto al fatto che esso non prende in considerazione alcun impedimento alla possibilità che il cammino ritorni su una posizione già precedentemente visitata, mentre è necessario tenere conto del fatto che i singoli segmenti monometrici si respingono a vicenda e sono mutuamente impenetrabili. Se la catena polimerica è perfettamente descritta da un cammino casuale, le sue dimensioni, descritte per es. dal raggio di girazione, risultano proporzionali alla potenza 1/2 del numero N di unità monometriche contenute nella catena, ma se si tiene conto del fatto che le unità monometriche si respingono a vicenda si calcola che il raggio di girazione dipende dalla potenza 3/5 di N.
La dimensione di una molecola di polimero è di conseguenza maggiore di quelle che si calcolerebbero per effetto del cammino casuale e la catena si dispone in una struttura più aperta.
Il tipo di simmetria che si osserva in una catena polimerica è simile a quella che si manifesta in un cluster di particelle, e viene definita ordine dilazionale; il suo aspetto caratteristico è rappresentato dal fatto che le proprietà del materiale non variano quando si opera un cambio di scala. Tuttavia la flessibilità di un polimero fornisce determinate proprietà che gli aggregati di particelle non possiedono. Questi ultimi risultano 'congelati' in configurazioni fisse, mentre le catene polimeriche sono libere di esplorare interi insiemi di direzioni casuali dei legami. Alla casualità di ciascuna configurazione di catena corrisponde una certa entropia, che può essere sfruttata come serbatoio di calore e lavoro. Una catena può subire grandi deformazioni sotto l'azione di perturbazioni deboli, non più grandi dell'energia termica kT, senza tuttavia patire effetti permanenti. Le dispersioni colloidali possono essere concentrate fino a raggiungere un contenuto di fase dispersa che risulta corrispondente alle condizioni 'di massimo impacchettamento'. Queste sono essenzialmente determinate da condizioni di tipo geometrico e dipendono dalle distribuzioni granulometriche delle particelle: per una dispersione monodispersa, in cui le particelle abbiano tutte le stesse dimensioni, le condizioni di massimo impacchettamento corrispondono a una frazione volumetrica di particelle pari a 0,63. Distribuzioni polidisperse rendono possibile il fare aumentare la frazione volumetrica che è corrispondente al massimo impacchettamento. I polimeri, invece, possono essere concentrati fino a frazioni volumetriche vicino all'unità. In assenza totale di solvente, le catene si interpenetrano e si attorcigliano, formando quelli che, con termine inglese, si chiamano entanglements, cosicché ogni catena interagisce con centinaia di altre catene. Ogni sollecitazione che è impartita sul sistema viene trasmessa e può produrre deformazioni grandi, ma reversibili, in ciascuna catena. Sono proprio queste deformazioni a produrre le forze di ritorno che agiscono nei materiali elastici. I polimeri fusi rispondono come materiali elastici quando le sollecitazioni sono veloci, se invece esse sono lente, le catene hanno un tempo sufficiente a liberarsi dai loro entanglements, dimenticandosi delle loro distorsioni originarie: in queste condizioni essi tendono a fluire, manifestando al contempo un comportamento prevalentemente viscoso, anziché elastico. Il moto di una catena polimerica, vincolato dalla presenza di tutte le altre catene a cui essa è attorcigliata, è stato descritto da un meccanismo denominato di reptazione. L'utilizzo di questo termine, che indica il movimento di molti rettili, in particolare dei serpenti, è spiegato dal fatto che il moto della catena polimerica ricorda proprio il movimento di un serpente che muove la sua lunga coda tra i fili d'erba, i quali a loro volta ben rappresentano le altre catene a cui il polimero è attorcigliato e che esso non può attraversare. La differenza tra un polimero fuso e un materiale elastico, ossia una gomma, è rappresentato dal fatto che in quest'ultimo gli entanglements sono permanenti, e vengono creati chimicamente da reazioni di reticolazione.
Il comportamento reologico delle dispersioni colloidali è, se possibile, più complesso di quello dei polimeri, per cui esiste carenza di modelli generali in grado di descriverlo. A. Einstein nel 1906 calcolò la dissipazione viscosa prodotta dal flusso intorno a una sfera singola, e ricavò un'equazione per il calcolo della viscosità di una dispersione, lineare nella frazione volumetrica φ delle particelle. Tale equazione però vale nel caso di sistemi diluitissimi, per valori di φ inferiori al 3%. In queste condizioni, inoltre, le dispersioni si comportano da fluidi newtoniani, in quanto la loro viscosità non dipende né dalla velocità di taglio cui sono sottoposte, né dal tempo in cui tali sollecitazioni vengono applicate. Al crescere della frazione volumetrica delle particelle le cose si complicano, nel senso che le dispersioni cominciano a manifestare comportamenti non newtoniani ed emergono dipendenze non lineari da φ. La viscosità cresce esponenzialmente a mano mano che φ si avvicina alla frazione di massimo impacchettamento.
A parità di frazione volumetrica le dispersioni flocculate hanno viscosità più alte delle dispersioni stabili, perché gli aggregati schermano il flusso, che tende a circondarli piuttosto che ad attraversarli. Tuttavia i flocculi, se sono reversibili, tendono a rompersi sotto l'azione delle sollecitazioni e la viscosità tende quindi a riportarsi al valore che competerebbe alla dispersione stabile. Quando i clusters di particelle assumono dimensioni tali da pervadere l'intera dispersione, questa presenta proprietà elastiche, oltre a manifestare una certa resistenza allo scorrimento, associata alla sollecitazione necessaria a rompere questa struttura diffusa.
È interessante sottolineare che, se la forza ionica della fase continua è molto bassa, anche le dispersioni stabilizzate elettrostaticamente possono assumere un ordine pseudocristallino per effetto delle forze repulsive che tendono a bloccare le particelle su posizioni fisse: queste strutture si chiamano cristalli colloidali, e manifestano un modulo elastico piuttosto elevato. Quando un cristallo colloidale viene fatto fluire, imponendo una sollecitazione superiore alla sua resistenza allo scorrimento, la sua struttura si riarrangia in modo da poter accogliere una deformazione continua.
Spesso le dispersioni colloidali, a velocità di taglio abbastanza elevate, si comportano da fluidi dilatanti, la cui viscosità aumenta al crescere della velocità di taglio. Questo tipo di fenomeno è associato alla formazione di strutture bidimensionali a strati, che sono comunque piuttosto instabili e tendono facilmente a essere distrutte.
Esistono molti aspetti e fenomeni attinenti alla reologia della m. s. che non sono ancora stati completamente compresi. Tra questi vale la pena citare la riduzione dell'attrito viscoso, per cui poche parti per milione di polimero aggiunte a un liquido sono in grado di alterare in maniera molto significativa il modo in cui questo scorre in un tubo, ritardando la dissipazione che si verifica vicino alle pareti in condizioni di flusso turbolento. Sebbene questo effetto sia ampiamente sfruttato nella pratica, non è stato ancora pienamente compreso, ed è oggetto di intenso studio in letteratura.
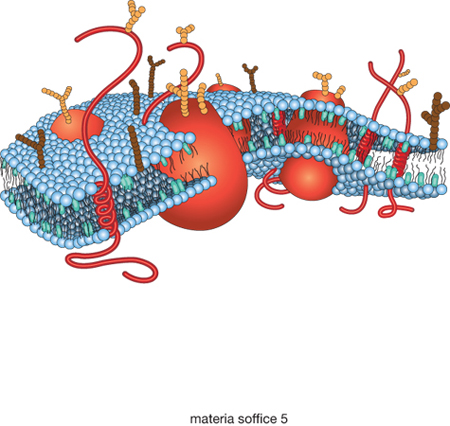
È già stata sottolineata l'importanza rivestita dalla m. s. nelle scienze della vita. Tradizionalmente lo studio della m. s. si occupava in gran parte di sistemi di origine biologica. La maggior parte delle colle e delle pitture derivava da prodotti di origine naturale, così come gran parte delle fibre. Dalla seconda metà del 20° sec. in poi, la strada della scienza dei colloidi e dei polimeri e quella della biologia si sono progressivamente separate. Da un lato infatti si è assistito all'invenzione dei polimeri sintetici e allo sviluppo massiccio dell'industria della plastica; dall'altro la scoperta del codice genetico e lo sviluppo della cristallografia delle proteine hanno aperto la strada alla nascita della biologia molecolare, che ha conseguito straordinari successi nel corso degli ultimi decenni. Tuttavia la biologia molecolare e lo studio della m. s. hanno ancora molti temi in comune. Basti notare che alcuni dei componenti chiave della biologia molecolare, come per es. gli acidi nucleici, le proteine e i polisaccaridi, sono polimeri. Inoltre molte delle più importanti biomembrane, come per es. i fosfolipidi, sono formate dall'assemblaggio di molecole anfifiliche, nelle quali due catene idrocarburiche si attaccano a un unico gruppo polare di testa. Questa tipologia di struttura favorisce la formazione di bistrati, anche se le biomembrane in realtà sono costituite da miscele complesse di fosfolipidi e altre molecole anfifiliche, come è possibile vedere in fig. 4, che mostra un diagramma schematico della membrana di una cellula eucariota. È ormai riconosciuto che biomembrane formate da lipidi sintetizzati per via abiotica ricoprirono un ruolo importantissimo all'origine della vita, dal momento che esse permisero di compartimentalizzare le prime reazioni biochimiche, costituendo così i prototipi delle cellule. Le biomembrane sono evolute a un grado di sofisticazione ben maggiore, ma il loro comportamento sottostà ai medesimi principi fisici che permisero la formazione di quei loro importantissimi progenitori.
bibliografia
I.W. Hamley, Introduction to soft matter, Chichester-New York 2000; R.A.L. Jones, Soft condensed matter, Oxford-New York 2002; T.A. Witten, P.A. Pincus, Structured fluids, Oxford-New York 2004.