
Creutzfeldt-Jakob, malattia di
Creutzfeldt-Jakob, malattia di
Patologia neurologica (in sigla MCJ) a esito fatale appartenente alle encefalopatie spongiformi trasmissibili (EST), o malattie da prioni dell’uomo, causate da un agente infettivo (prione) le cui caratteristiche strutturali sono in gran parte sconosciute, ma che presenta alcune particolarità: è resistente alle procedure comunemente impiegate per rimuovere o inattivare microrganismi noti, non stimola una classica risposta immunitaria e induce nell’ospite un lungo tempo di incubazione che può variare da alcuni anni a decenni. La MCJ è anche considerata malattia da alterata conformazione proteica, insieme alla malattia di Alzheimer, a quella di Parkinson, alla sclerosi laterale amiotrofica e alla corea di Huntington. Nella MCJ, la proteina prionica cellulare (PRPC) assume una o più conformazioni patologiche (PRPEST) che si accumulano nel cervello e, in misura minore, nei tessuti linforeticolari dei soggetti infetti.
Malattia di Creutzfeldt-Jakob sporadica
La MCJ sporadica è diffusa in tutto il mondo con un’incidenza annua che varia tra 1 e 2 casi per milione di persone. Colpisce di preferenza soggetti tra i 60 e gli 80 anni con una durata complessiva che varia tra i 3 e i 6 mesi. L’esordio della malattia può essere insidioso, con disturbi cognitivi o comportamentali, allucinazioni visive o disturbi del coordinamento motorio (forme atassiche). Più raramente, la malattia insorge in modo acuto simulando un accidente vascolare cerebrale. La malattia progredisce piuttosto rapidamente e nel giro di poche settimane il paziente presenta una grave compromissione delle funzioni superiori (demenza), segni cerebellari, mioclonie diffuse, fino a raggiungere uno stadio di quasi totale immobilità e mutismo (mutismo acinetico). La diagnosi clinica è molto difficile nelle prime settimane di malattia ma diventa relativamente semplice con il rapido progredire della malattia e con la comparsa del mioclono (➔ movimento, disturbi del). Alcuni esami strumentali, come l’elettroencefalogramma (EEG), la risonanza magnetica (RM) del cranio e la presenza della proteina 14-3-3 nel liquido cefalorachidiano sono di grande utilità per formulare una corretta diagnosi clinica. L’EEG mostra in circa il 60% dei casi un ritmo pseudo-periodico molto caratteristico e la RM del cranio una iperintensità bilaterale dei nuclei della base e spesso anche della corteccia cerebrale. Non è noto alcun fattore di rischio responsabile della MCJ sporadica se non una debole predisposizione genetica legata al sito polimorfico del gene della proteina prionica (PRNP) al codone 129. Il polimorfismo in posizione 129 influenza la suscettibilità alla malattia e il suo decorso clinico. Nella popolazione generale questo sito polimorfico produce due varianti alleliche, che codificano gli aminoacidi metionina e valina. La distribuzione del genotipo al codone 129 nelle popolazioni caucasiche dimostra che circa il 40% degli individui è omozigote per metionina, il 50% è eterozigote (metionina/ valina) e il restante 10% circa è omozigote per valina. Questi valori differiscono significativamente da quelli che si ritrovano tra i pazienti affetti da MCJ sporadica, nei quali il 70÷80% dei casi è omozigote per metionina. Recentemente è stato riscontrata una maggior frequenza di interventi chirurgici nei pazienti con MCJ sporadica rispetto alla popolazione di controllo. Non vi sono al momento terapie efficaci per la MCJ, anche se alcuni trial clinici con sostanze che dovrebbero essere in grado di bloccare l’alterata conformazione della PrPc in PrPEST sono in corso negli Stati Uniti, nel Regno Unito e in Italia.
Malattia di Creutzfeldt-Jakob iatrogena
Questa forma di MCJ è causata dalla trasmissione dell’agente infettivo attraverso l’utilizzo di ormoni ipofisari estratti da tessuti cadaverici (per es., l’ormone della crescita) o impianti di dura madre prelevati da pazienti deceduti per MCJ sporadica. Alcuni casi di MCJ iatrogena sono stati descritti anche in seguito all’utilizzo di ferri neurochirurgici utilizzati precedentemente in pazienti con MCJ sporadica e non adeguatamente sterilizzati. La sintomatologia clinica della MCJ iatrogena è simile a quella della forma sporadica.
Malattia di Creutzfeldt-Jakob variante
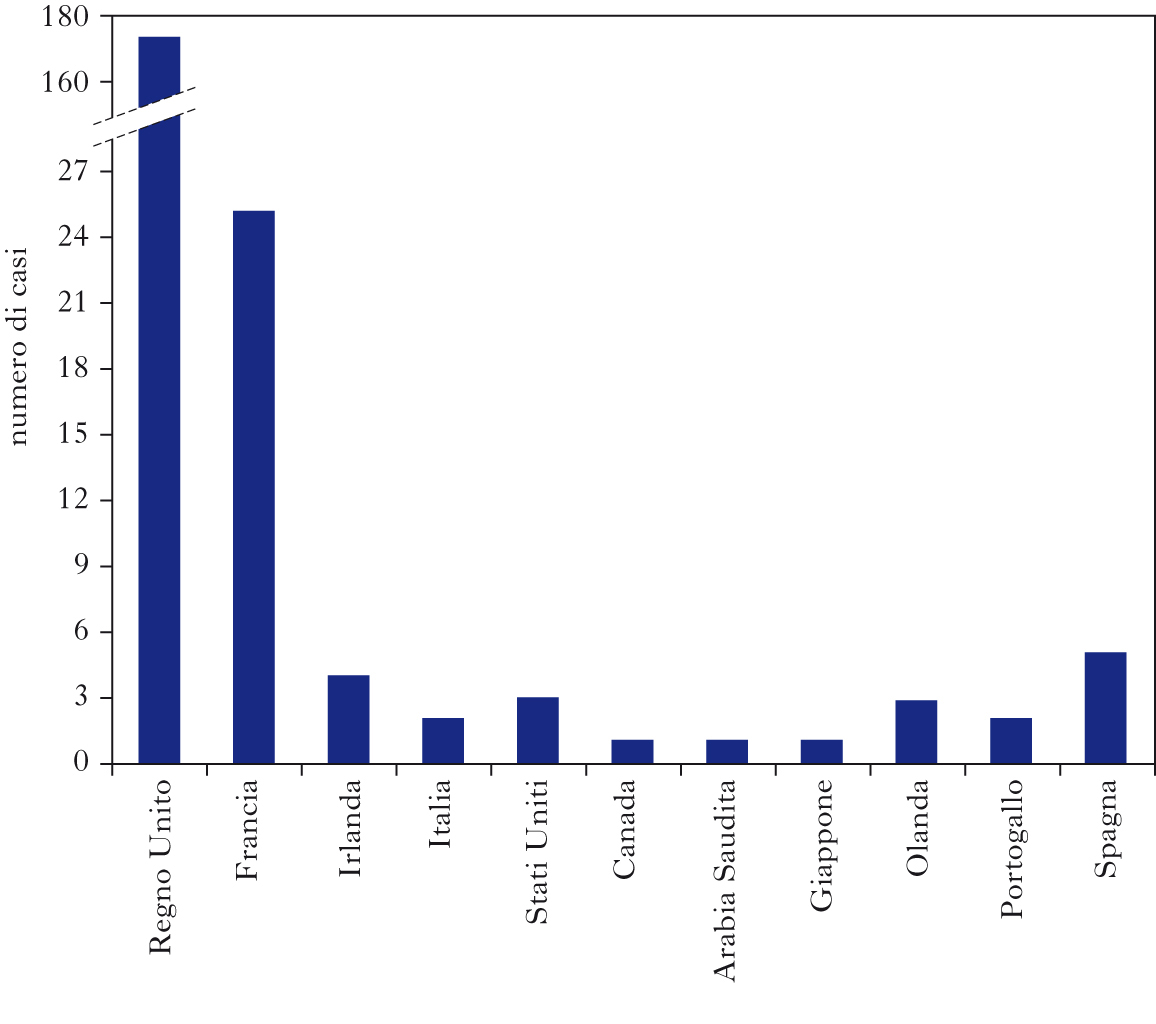
La MCJ variante è causata dall’agente dell’encefalopatia spongiforme bovina, in seguito a consumo di cibo infetto, e colpisce prevalentemente soggetti tra i 15 e i 30 anni. A marzo 2010 sono stati descritti 219 casi, in prevalenza nel Regno Unito (169) e in Francia (25). La MCJ variante si discosta dalla MCJ sporadica per la durata della malattia, superiore ai sei mesi, e per le caratteristiche cliniche di esordio di tipo psichiatrico (depressione, ansietà, apatia, illusioni). La sintomatologia evolve nei mesi successivi con un’atassia della marcia, disturbi sensoriali di tipo dolorifico, movimenti involontari, progressivo deterioramento intellettivo e mutismo acinetico, con un quadro neurologico franco, sostanzialmente non dissimile dalla forma sporadica. Fondamentale per la diagnosi clinica di MCJ variante è l’esecuzione della RM del cranio che mostra un’iperintensità bilaterale di segnale a livello del pulvinar. La presenza della proteina liquorale 14-3-3 è incostante. Quando la sintomatologia clinica e gli esami strumentali non permettono di distinguere la variante MCJ da altre forme di MCJ, si ricorre alla biopsia tonsillare per identificare, mediante tecniche immunochimiche, la presenza di PRPEST che è presente nei tessuti linforeticolari dei soli pazienti affetti da MCJ variante. La malattia può anche essere trasmessa da uomo a uomo attraverso trasfusioni di sangue o, probabilmente, in seguito a terapia con alcuni plasma-derivati (fattore VIII).
Malattia di Creutzfeldt-Jakob genetica
La MCJ genetica è legata a mutazioni del gene PRNP. La sintomatologia e la durata della malattia in questi pazienti sono generalmente simili a quelle della MCJ sporadica. Oltre alla MCJ genetica, esistono due altre rare forme di EST causate da mutazioni del gene PRNP: l’insonnia fatale familiare (➔), che si manifesta con gravi alterazioni del sonno e del sistema nervoso vegetativo e la sindrome di Gerstmann- Sträussler-Scheinker, nella quale i pazienti sviluppano principalmente disturbi di tipo cerebellare, con una durata di malattia di oltre 3 anni.