
La grande scienza. Le basi genetiche ed epigenetiche del cancro
La grande scienza. Le basi genetiche ed epigenetiche del cancro
Le basi genetiche ed epigenetiche del cancro
Il cancro si presenta in un'ampia varietà di tessuti in forme notevolmente differenti, ognuna delle quali implica comunque un'alterazione genetica. L'osservazione che nelle cellule sia la perdita sia l'acquisizione di informazione genetica possano contribuire direttamente all'insorgenza del cancro fu effettuata per la prima volta circa un secolo fa, ma non ricevette ampi consensi. Tuttavia, negli ultimi quindici anni numerosi studi di epidemiologia, di citogenetica e di genetica molecolare hanno fornito numerose prove a supporto di questa ipotesi.
L'epidemiologia genetica del cancro, spesso condotta nell'ambito di studi sulle famiglie, fornisce una prima valutazione del grado di associazione tra mutazioni ereditarie e predisposizione alla malattia. Questi studi hanno mostrato che il cancro si manifesta con molta più frequenza, o tende a 'raggrupparsi', in alcune famiglie rispetto ad altre. Ciò è stato dimostrato per molte forme comuni di cancro, compreso quello della mammella, dell'ovaio, del colon, della prostata, del cervello e del polmone. Il modello che mostra il rischio crescente nell'ambito di queste famiglie indica che il 'raggruppamento' è dovuto a una predisposizione ereditaria al cancro, piuttosto che alla comune esposizione ai danni provocati da agenti ambientali. Ciò è illustrato da due esempi ben documentati relativi al cancro del seno e a quello della prostata. In un ampio studio sul cancro familiare della prostata, l'aumento dei rischi per un maschio con uno, due o tre parenti affetti era, rispettivamente, di 2,2, 4,9, 10,9. Il rischio aumentava ulteriormente (10-11 volte) in individui con due parenti colpiti dalla malattia, di cui uno aveva ricevuto la diagnosi prima dei quarant'anni. Anche studi sul cancro ereditario della mammella hanno mostrato un aumento di rischio in individui con un parente di primo grado (1,8), o più d'uno (3,3), affetto dalla stessa malattia. Come per il cancro della prostata, un ulteriore aumento del rischio di cancro della mammella è stato osservato quando uno dei parenti colpiti dalla malattia aveva ricevuto la diagnosi in giovane età (trenta o quarant'anni). Questi risultati indicano che l'entità del rischio è funzione del numero dei parenti colpiti, e il rischio maggiore è per quei soggetti con più parenti colpiti dalla malattia in giovane età. Tali evidenze sono a favore dell'ipotesi di una trasmissione ereditaria di un difetto che predispone al cancro.
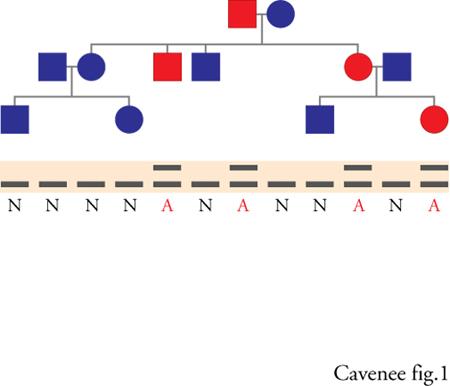
L'alta frequenza con cui alcuni tipi di tumore si manifestano nell'ambito di certe famiglie suggerisce una componente ereditaria nella eziologia del cancro, ma di per sé non fornisce alcun indizio circa il modo in cui il carattere genetico della malattia viene trasmesso. La modalità di trasmissione del carattere canceroso nell'ambito di ognuna di queste famiglie (per un ipotetico 'albero genealogico della malattia', fig. 1, in alto) si può dedurre confrontando i dati relativi alle singole famiglie con quelli previsti dai diversi modelli di trasmissione genica della malattia. Il modello che meglio spiega la maggior parte dei raggruppamenti familiari (fig. 2) della predisposizione al cancro, esemplificato dall'albero genealogico della malattia nella fig. 1, è detto 'eredità autosomica dominante'. In questo modello l'ereditarietà della malattia non è legata al sesso; può quindi essere trasmessa da entrambi i genitori e implica la trasmissione di uno o più geni, la cui presenza è sufficiente a causare l'insorgere della malattia. Questa modalità di trasmissione autosomica dominante prevede che in media il 50% dei figli di un genitore colpito dal cancro svilupperà la malattia. Da un punto di vista epidemiologico, considerazioni sulla frequenza del raggruppamento familiare di predisposizione al cancro, secondo il modello della trasmissione autosomica dominante, portano a concludere che il 5-10% di tutti i tipi di cancro studiati ha come causa l'ereditarietà di uno o più geni dominanti; gli altri tipi vengono definiti sporadici.
Oltre al raggruppamento familiare di alcuni tipi comuni di cancro, due ulteriori osservazioni cliniche forniscono un valido supporto epidemiologico all'opinione che il cancro abbia un'origine genetica. In primo luogo, alcuni individui e le loro famiglie presentano una trasmissione autosomica dominante di predisposizione non solo per un singolo tipo, come descritto sopra, ma per molteplici tipi di cancro che si manifestano in modo indipendente in differenti organi e tessuti del corpo. In secondo luogo, in individui che presentano una varietà di difetti dello sviluppo in differenti organi si manifestano spesso anche rare forme tumorali specifiche. Attraverso una valutazione statistica si può desumere che l'insorgenza di più tumori indipendenti, o la frequente associazione di difetti nello sviluppo con tumori che di per sé sono rari nella popolazione, è così improbabile da suggerire una correlazione eziologica.
Gli argomenti statistici e i modelli di trasmissione derivati dagli studi di epidemiologia dei tumori forniscono una base su cui condurre la ricerca dei geni responsabili della malattia attraverso l'analisi genetica dell'associazione (linkage). Le tecniche di citogenetica e di genetica molecolare vengono utilizzate per identificare un'associazione statisticamente rilevante tra il fenotipo della malattia e specifici difetti genetici (fig. 1). Questa ricerca inizia con un'analisi microscopica dei cromosomi delle cellule normali e tumorali di individui con una predisposizione ereditaria al cancro.
I cromosomi in metafase di cellule che vanno incontro alla mitosi vengono colorati in modo che ciascuno di essi assuma una caratteristica serie di bande chiare e scure, una specie di codice a barre che serve a identificare ogni singolo cromosoma. Le deviazioni dal normale modello di distribuzione delle bande nei cromosomi, riscontrate in tutte le cellule di individui malati, e che sono assenti nei parenti non affetti dalla malattia, circoscrivono la ricerca del gene tumorale alla regione cromosomica anomala. Purtroppo, le regioni cromosomiche identificate con le tecniche citogenetiche sono quasi sempre così grandi da contenere centinaia o anche migliaia di geni, dei quali solamente uno o pochi sono i veri responsabili del fenotipo canceroso.
Gli strumenti della genetica molecolare, e in particolare la combinazione della tecnica di ibridazione nota come Southern blot e della reazione a catena della polimerasi (PCR, polymerase chain reaction), sono quindi utilizzati per restringere la regione sospetta a un segmento di DNA contenente soltanto pochi geni che possono essere analizzati direttamente per evidenziare i possibili difetti negli individui colpiti dalla malattia. Il procedimento che va dall'analisi epidemiologica alla citogenetica e la genetica molecolare hanno portato sinora all'isolamento di più di venti geni che, qualora ereditati nella forma mutata, predispongono l'individuo allo sviluppo di particolari tipi di tumori.
Il retinoblastoma: un prototipo di cancro ereditario e sporadico
L'apparente trasmissione dominante del carattere genetico del cancro, descritta precedentemente, risulta essere paradossale alla luce di tre elementi: (1) cellule ibride ottenute sperimentalmente dalla fusione di cellule tumorali fortemente maligne con cellule normali non sono solitamente tumorigene, e ciò suggerisce che il fenotipo normale sia dominante in presenza di mutazioni tumorigene. Inoltre, nei pochi casi in cui una cellula ibrida diventa tumorigena, essa ha perduto cromosomi specifici originariamente forniti dalla cellula normale, e ciò implica che il fattore responsabile del fenotipo tumorale non è l'acquisizione di un carattere tumorale dominante, ma una perdita di specifici cromosomi; (2) se una singola mutazione fosse sufficiente a provocare un tumore, ci dovremmo aspettare che famiglie che segregano forme autosomiche dominanti di cancro non presentino alcun tipo di tessuto normale nell'organo malato. Questa aspettativa è in contrasto diretto con la descrizione clinica di tumori che presentano lesioni focalizzate circondate da tessuti normali e funzionanti dello stesso organo; (3) analisi epidemiologiche di forme sporadiche e familiari di diversi tipi di cancro umano, in contrasto con la trasmissione autosomica dominante, hanno indicato che la conversione di una cellula normale in una tumorale richiede una serie di eventi (Knudson 1971).
Questi paradossi sono stati infine risolti attraverso lo studio del retinoblastoma, un tumore che è servito come prototipo per comprendere la genetica della maggior parte dei tumori dell'uomo ereditari e sporadici. Il retinoblastoma è un tumore relativamente raro (10 ogni 20.000 nascite) che colpisce i bambini e che si manifesta sia in forma sporadica sia in forma ereditaria autosomica dominante. Sulla base di soli dati statistici derivanti da osservazioni cliniche ed epidemiologiche, sono state tratte molte conclusioni importanti circa la natura degli eventi che conducono alla formazione del retinoblastoma (Knudson 1971). In primo luogo, la mutazione ereditata non è da sola sufficiente a causare il tumore dato che vi sono almeno 107 cellule (retinoblasti) che rappresentano potenziali bersagli per i retinoblastomi, ognuna portatrice della mutazione ereditata, mentre in media si formano solo tre tumori indipendenti in ogni individuo colpito dalla malattia. Ciò suggerisce inoltre che a livello genetico le mutazioni che causano il retinoblastoma possono essere recessive, piuttosto che dominanti come suggerirebbe il modello di trasmissione. È stato ipotizzato che i tumori ereditari originino da una prima mutazione a livello della linea germinale, seguita da una seconda mutazione in una cellula somatica. La frequenza delle mutazioni somatiche è simile sia nei tumori sporadici sia in quelli ereditari, sebbene i primi necessitino di due mutazioni somatiche, ognuna nello stesso retinoblasto, per la formazione del tumore. In completo accordo con questo è l'osservazione che i casi ereditari si verificano solitamente in età precoce, sono spesso bilaterali e sviluppano tumori multipli, mentre al contrario i casi sporadici sono sempre unilaterali e sviluppano singoli tumori. Poiché nei casi ereditari vi è una bassa probabilità che una seconda mutazione somatica non si verifichi mai, circa il 5% dei portatori non sviluppa alcun tumore. La natura dei due bersagli della mutazione del genoma era sconosciuta al momento di tali osservazioni cliniche, ma la citogenetica e la genetica molecolare hanno consentito di rispondere a questo quesito e hanno offerto un approccio generale allo studio di altri tipi di cancro umano. L'analisi del bandeggio cromosomico in pazienti affetti da retinoblastomi ereditari e sporadici ha rivelato una delezione del cromosoma 13q14 (cromosoma 13, braccio q o lungo, banda uno-quattro), suggerendo che il gene per il retinoblastoma (Rb) è localizzato in qualche parte di questa regione. Il DNA estratto da cellule di tumori ereditari è stato poi analizzato con sonde di DNA clonato (chiamate DNA markers) che potevano distinguere le due copie, o alleli, del cromosoma 13 in ciascuna cellula. È stato riscontrato che nelle cellule tumorali la regione contenente il gene sospetto Rb sul cromosoma 13 era presente solo in una forma mutata. Questa conversione da uno stato di eterozigosi a uno di omozigosi per la mutazione è stata chiamata 'perdita di eterozigosi' (LOH, loss of heterozygosity) e costituirebbe il secondo evento necessario per la formazione di tumori nei casi ereditari (Cavenee et al. 1983). Inoltre la LOH sul cromosoma 13q14 si verificava anche nei casi di retinoblastoma sporadico. Questi dati hanno fornito un supporto notevole all'ipotesi secondo la quale la formazione tumorale del retinoblastoma potrebbe verificarsi in seguito allo 'smascheramento' di un difetto genetico recessivo. La scoperta che, nell'uomo, la LOH si verifica in altre forme tumorali ereditarie e nella maggior parte dei tumori sporadici (Rodriguez et al. 1994) ha dato il via, contemporaneamente, alla genetica delle cellule cancerose somatiche e a quella del cancro ereditario. È stato infine identificato il gene responsabile del retinoblastoma: le sue funzioni sono state determinate grazie all'identificazione della regione cromosomica 13q14 con la LOH più consistente nel DNA tumorale. L''ipotesi dei due eventi' (two hit hypothesis) si è rivelata valida a livello genetico per i retinoblastomi ereditari e sporadici, ma, come si vedrà nel prossimo paragrafo, è stata altrettanto valida anche per spiegare la formazione di quasi tutti gli altri tipi di tumore, sia ereditari sia sporadici.
Altri geni soppressori tumorali
Geni identificati dalla predisposizione familiare
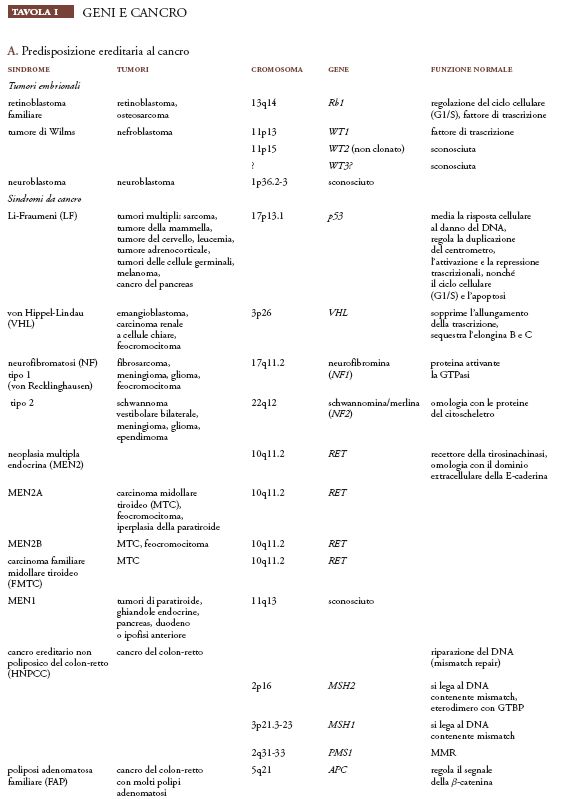

Attraverso l'associazione genetica e gli studi della LOH nel DNA delle cellule tumorali, sono stati identificati più di venti geni che, se ereditati in forme mutanti, causano la predisposizione a uno specifico tipo di cancro. In maniera simile al retinoblastoma, ognuna di queste predisposizioni al cancro è trasmessa come un carattere autosomico dominante e, a livello genico, tutte eccetto una (oncogene RET) sono dovute a un allele mutante recessivo ereditato che viene smascherato da una mutazione successiva o dalla perdita della rimanente copia normale. Questi geni sono detti 'soppressori tumorali' perché la perdita delle loro normali funzioni di controllo della crescita conduce direttamente o indirettamente alla formazione tumorale. Considerando esclusivamente i tumori ereditari, è di 50 ca. il numero totale di potenziali geni soppressori tumorali umani (Tav. Ia e Ib.A), dato che questo è il numero di differenti tipi di tumori ereditari descritti mediante studi di carattere epidemiologico e clinico (Mulvihill 1977).
Il tumore di Wilms del rene è stato il secondo tumore embrionale dei bambini a mostrare il coinvolgimento della LOH nella sua eziologia. Questo tumore può manifestarsi da solo o in combinazione con uno dei diversi disordini genetici nello sviluppo, tra cui l'aniridia sporadica o le sindromi WAGR, di Denys-Drash o di Beckwith-Wiedemann. A differenza del retinoblastoma, almeno tre diversi geni possono predisporre al tumore di Wilms, dei quali solo uno, il WT1, è stato clonato.
Nonostante le comuni modalità di trasmissione che si riscontrano tra i tumori ereditari, la percentuale di portatori del gene che, di fatto, sviluppa la malattia varia a seconda dei diversi tipi di cancro. Per esempio, il 90-95% degli individui che ereditano le mutazioni del gene Rb sviluppa il retinoblastoma (Knudson 1971); il 5-10% dei portatori che non lo sviluppa sembra riflettere la piccola possibilità, statisticamente ben definita, che non avvenga una seconda mutazione in una cellula somatica. Al contrario, la trasmissione ereditaria del tumore di Wilms mostra una 'penetranza' incompleta, essendo non penetrante in alcune generazioni, ma molto penetrante in quelle successive. Ciò potrebbe essere dovuto a modificazioni epigenetiche del carattere della malattia, dato che nei tumori di Wilms ereditari e sporadici vengono perduti prevalentemente gli alleli materni. Individui con le mutazioni BRCA1 hanno una penetranza specifica del tumore sia per il tumore della mammella (80%) sia per quello dell'ovaio (40%). La penetranza della malattia in individui con mutazioni BRCA1 è modificata da un secondo gene, HRAS1. I portatori di BRCA1 che portano un allele HRAS1, raro nella maggior parte della popolazione, hanno probabilità 2,1 volte maggiori di sviluppare il tumore dell'ovaio rispetto a quelli in cui l'allele HRAS1 è il più comune. Il significato funzionale di tale allele raro è sconosciuto, ma questa è la prima osservazione che mostra l'effetto di un modificatore sulla penetranza di una sindrome di cancro ereditario.
Sebbene l'inattivazione di molti geni soppressori di tumori richieda una mutazione che causa la perdita funzionale in entrambe le coppie del gene normale, cellule eterozigoti per forme mutanti di altri geni soppressori tumorali, come p53 o WT1, potrebbero avere un vantaggio nella crescita rispetto a cellule normali grazie a un comportamento dominante negativo. La proteina p53 è attiva in forma tetramerica ed è resa inattiva dall'incorporazione di uno o più monomeri di p53 mutante normalmente sviluppati che mantengono la capacità di legarsi con altre subunità e formare oligomeri. Similmente, nel tumore di Wilms una mutazione della proteina WT1, che risulta alterata per il legame con il DNA, contrasta l'attività della normale WT1 formando oligomeri con questa (Moffett et al. 1995). In tal modo, cellule eterozigoti per i mutanti p53 o WT1 possono avere un vantaggio nella crescita, se vengono espressi sia il gene mutante sia quello normale. Oltre alla perdita di funzione o all'attività dominante negativa, p53 può acquisire nuove proprietà attraverso mutazioni di acquisto di funzione che determinano un aumento delle proprietà tumorigene nelle cellule mancanti di p53 normale.
Il gene RET, che è coinvolto nella neoplasia endocrina multipla di tipo 2 (MEN2, multiple endocrine neoplasia type 2) e nel carcinoma familiare midollare tiroideo (FMTC, familial medullary thyroid carcinoma), è l'unico che predispone al cancro noto per non essere un gene soppressore tumorale (Mak e Ponder 1996). MEN2 e FMTC sono trasmessi in modo autosomico dominante, ma la trasmissione uniparentale di un solo gene RET mutato può essere sufficiente a provocare la malattia, anche in presenza di un allele RET normale. L'analisi funzionale delle proteine RET mutate ha rivelato un'attivazione costitutiva dell'attività intrinseca della tirosinachinasi e un'alterata specificità peptide substrato. Quindi il gene RET sembra seguire un modello di oncogene dominante, piuttosto che una perdita della funzione di soppressore. È tuttavia paradossale che i tumori associati al gene RET siano di natura focale alla stregua di quelli ereditari associati ai geni soppressori, invece di coinvolgere tutte le cellule dei tessuti endocrini colpiti, come ci si potrebbe attendere da un allele mutante dominante. È dunque possibile che sia necessaria una seconda alterazione genetica o epigenetica per la formazione del tumore, come è inoltre suggerito dalla presenza di mutazioni sia somatiche sia germinali del gene RET nei tumori di numerosi individui affetti da FMTC. Alterazioni del gene RET sono coinvolte anche nei tumori sporadici, inclusi i carcinomi midollari e della tiroide. Un'alta incidenza di alterazioni del gene RET è stata registrata nei tumori della tiroide di bambini della Bielorussia, nella ex Unione Sovietica, molti anni dopo l'incidente nucleare di Černobyl del 1986, suggerendo l'ipotesi di alterazioni del gene RET indotte dalle radiazioni.
Diversi geni soppressori tumorali, identificati grazie allo studio della predisposizione ereditaria al cancro, sono coinvolti anche in uno spettro piuttosto ampio di tumori sporadici. Ciò è ben illustrato da tre geni, Rb, p53 e p16/pCDKN2. A causa del numero limitato di forme tumorali riscontrate in pazienti con retinoblastoma ereditario, è stato alquanto sorprendente scoprire che il gene del retinoblastoma è alterato in molti tumori sporadici di individui adulti, inclusi il cancro del polmone, della mammella, della prostata, del pancreas, del cervello, nei sarcomi dei tessuti molli e ossei, nelle leucemie e nei linfomi. Per molti di questi tumori sporadici la LOH del gene Rb sembra essere coinvolta nella progressione del tumore piuttosto che nel suo inizio. Le mutazioni del gene p53 nella linea germinale predispongono a molti tipi di tumori nella sindrome di Li-Fraumeni, oppure possono predisporre a un singolo tipo di tumore (per es., i tumori del cervello). Le mutazioni del gene p53 nella linea somatica sono le alterazioni genetiche più frequentemente osservate nei tumori sporadici; infatti si verificano approssimativamente nel 50-60% di tutti i tumori esaminati, inclusi quelli della mammella, del colon, del fegato, del polmone e del cervello. Le mutazioni del gene p53 insorgono negli stadi precoci dei tumori del cervello e del polmone, ma molto più tardi nel tumore del colon (Fearon et al. 1990), suggerendo che la mutazione p53 potrebbe essere coinvolta sia nella fase iniziale sia nella progressione dei tumori. Le alterazioni del gene p16, presenti nel melanoma familiare, sono frequenti anche nei tumori sporadici, inclusi quelli del pancreas, della vescica, della testa e del collo, delle cellule dei reni, del polmone, della mammella, della prostata e del cervello. La diversità delle forme tumorali che coinvolgono le mutazioni nei geni Rb, p53 o p16 è probabilmente connessa alla manifestazione ubiquitaria di questi geni nelle cellule normali. I geni BRCA1 (Miki et al. 1994) e BRCA2 per i tumori ereditari, rispettivamente della mammella e di mammella e ovaie, non sono in genere mutati nei tumori sporadici della mammella, costituendo in tal modo due importanti eccezioni al doppio coinvolgimento di geni in tumori sia ereditari sia sporadici. Per altri geni soppressori tumorali il coinvolgimento esclusivo in un tipo particolare di tumore (per es., il tumore di Wilms) può essere spiegato da un'espressione limitata nel tempo e nel tessuto (per es., l'espressione di WT1 durante lo sviluppo del rene e dei tessuti delle gonadi). In alcuni casi le modalità di espressione di un gene soppressore tumorale nei tessuti normali possono quindi fornire indizi sul tipo di tumore che può essere causato dalla sua mutazione.
Oltre alla segregazione con la predisposizione al cancro, la perdita di funzione di geni soppressori tumorali è testata sperimentalmente mediante tecniche di transfezione e delezione. Il gene Rb normale inibisce la crescita delle cellule tumorali se inserito, tramite transfezione, in cellule tumorali in coltura mancanti di copie funzionali di Rb. Topi che presentano una eliminazione, o knock out, mirata di una copia del gene Rb mostrano una forte predisposizione allo sviluppo del tumore, anche se non sviluppano retinoblastomi. Non è ancora chiaro se questo sia collegato al minor numero di retinoblasti presenti nei topi (Knudson 1993), o sia semplicemente inerente alle differenze tra specie.
A livello molecolare si hanno conoscenze approfondite sulla funzione della proteina Rb nella regolazione della crescita cellulare (Hunter e Pines 1994). Rb è una fosfoproteina nucleare 110 kDa che nella forma non fosforilata lega il fattore di trascrizione E2F e inibisce la progressione del ciclo cellulare nel punto di controllo G1/S. L'interazione con complessi di ciclina D1-CDK4 (chinasi 4 ciclina-dipendente) o di ciclina D1-CDK6 inattiva Rb mediante fosforilazione, promuovendo così la transizione attraverso la fase G1 del ciclo cellulare. I regolatori di Rb sono a loro volta controllati, aggiungendo ulteriori livelli di complessità al controllo del ciclo cellulare mediato da Rb. Almeno due inibitori di CDK, p15 e p16, possono legarsi e inibire CDK4 e CDK6. Mutazioni del gene p16 sono responsabili della predisposizione ereditaria al melanoma (Tav. Ia e Ib.A), fornendo così, inaspettatamente, un meccanismo comune per l'origine di due neoplasie ereditarie molto differenti. Nei tumori sporadici, alterazioni genetiche o epigenetiche di uno di almeno 6 di questi componenti della via di Rb può eliminare l'inibizione della crescita cellulare di Rb e contribuire al carattere maligno, sia attraverso una perdita di funzione (Rb, p16, p15) sia mediante un'acquisizione di funzione/sovrapproduzione (CDK4, CDK6, ciclina D1), sottolineando così l'importanza di questa via nel cancro umano. Rb inibisce la funzione della proteina ABL e interagisce anche con diverse proteine scoperte recentemente, RBP1, RBP2, BRG1 e RIZ, ma i ruoli specifici di queste interazioni nelle cellule normali e tumorali non sono stati ancora stabiliti.
È facile capire come la perdita di alcuni geni soppressori tumorali, come quelli che regolano negativamente il ciclo cellulare (Rb, p16, p15) o quelli che inibiscono i segnali di crescita positivi quali NF1 e APC, contribuisca direttamente allo sviluppo del tumore. La perdita di altri geni, quali MSH2, MSH1, PMS1 e PMS2 coinvolti nella riparazione di errati appaiamenti di basi del DNA (MMR, mismatch repair), richiede una spiegazione indiretta e quindi più complicata del perché causi la perdita del controllo della crescita e la predisposizione ai tumori. Una possibilità è che una riparazione inadeguata del DNA accresca la frequenza di mutazioni e alla fine conduca alla mutazione di un altro gene (o di altri geni) che esercita un effetto diretto sulla crescita o sulla divisione cellulare. Un potenziale gene effettore, che codifica il recettore di tipo II per il fattore trasformante di crescita β (TGF-β RII) è mutato nel suo tratto di poliadenina nel 90% dei tumori colon-rettali che mostrano, in coltura, instabilità genomica; in alcuni di essi è presente un gene MMR mutato. L'effetto inibitore della crescita da parte di TGF-β, che richiede entrambi i recettori di tipo I e tipo II, è abolito nella maggior parte dei tumori colon-rettali RER. Questi dati suggeriscono che le mutazioni TGF-β RII potrebbero avere qualche ruolo nella mediazione dell'effetto soppressore dei geni per la riparazione del DNA. Non è tuttavia ancora chiaro se il tipo di mutazioni di TGF-β RII identificate in questo studio sia sufficiente a produrre un tale effetto o se il gene TGF-β sia un bersaglio critico, dal momento che numerosi altri siti del genoma in queste cellule sono mutati con frequenze anche più alte. È comunque chiaro che l'alterazione genetica critica che predispone a questi tipi di tumori del colon è la perdita di funzione tramite mutazione ereditaria e la LOH di un gene soppressore tumorale con attività di riparazione MMR del DNA.
Geni identificati da instabilità cromosomica

Difetti nella riparazione del DNA sono altresì implicati nella forte predisposizione al cancro che accompagna quattro sindromi di instabilità cromosomica (Tav. I.B).
A differenza di altre forme di predisposizione al cancro, queste sindromi sono trasmesse in forma autosomica recessiva e sono di conseguenza assai rare: circa 1 caso su 300.000 per l'atassia-teleangectasia (AT), ossia da dieci a quindici volte meno del retinoblastoma. Sono stati clonati il gene responsabile dell'atassia-teleangectasia, sei dei sette geni responsabili di diversi gruppi di complementazione dello xeroderma pigmentoso (XP) e uno dei tre geni responsabili della sindrome di Fanconi (FA).
Un'aumentata predisposizione al cancro in individui omozigoti per un gene mutato è evidente in modo drammatico nei pazienti affetti da AT e XP. Rispetto alla popolazione generale, i pazienti affetti da AT presentano un incremento del rischio di sviluppare leucemia o linfoma, rispettivamente di 70 e di 250 volte. L'incidenza del carcinoma basale e di quello dell'epitelio a cellule squamose nei pazienti affetti da XP è ben 2000 volte maggiore rispetto a gruppi della stessa età nella popolazione generale. Pur essendo gli individui omozigoti rari, il numero di eterozigoti è ovviamente molto più alto. Numerosi studi epidemiologici suggeriscono che gli eterozigoti per il gene dell'AT, che si stima comprendano l'1% della popolazione generale, potrebbero avere la predisposizione a sviluppare il cancro, con un incremento del rischio generale di tre-quattro volte, e fino a cinque volte nel caso di tumori della mammella nelle donne eterozigoti per AT. Dal momento che la sensibilità degli eterozigoti per AT alle radiazioni è intermedia tra quella dei pazienti omozigoti e degli individui non affetti, l'utilizzazione della mammografia per diagnosticare il cancro della mammella è assolutamente da evitare negli eterozigoti per AT. È stato suggerito, sebbene non sia stato provato, che la sensibilità alle radiazioni e la predisposizione al cancro degli eterozigoti per AT potrebbe dipendere dal dosaggio genico (un solo gene AT normale). La recente clonazione del gene AT permetterà di verificare a livello molecolare queste interessanti scoperte epidemiologiche. Un'analisi preliminare del gene AT in 38 tumori della mammella sporadici scelti a caso, e nelle corrispondenti cellule normali del sangue, non ha rivelato un aumentato numero di eterozigoti per AT.
Geni identificati nei tumori sporadici

Oltre all'identificazione di geni soppressori tumorali tramite difetti genetici ereditati, molti geni candidati sono stati isolati tramite studi della LOH nei tumori sporadici (Tav. I.C).
Il numero sempre crescente di marcatori del DNA disponibili e lo sviluppo dell'analisi PCR multipla, cioè su più regioni contemporaneamente, basata sulla fluorescenza e assistita da computer, ha fortemente aumentato le possibilità applicative della LOH. Nuove metodiche di esplorazione del genoma, quali RLGS (restriction landmark genomic scanning) e RDA (representational difference analysis), potranno essere di aiuto nell'identificazione di questi geni. Dati sulle sequenze provenienti dal progetto 'genoma umano' faciliteranno senz'altro questi studi.
Meccanismi cromosomici responsabili dell'inattivazione dei geni soppressori tumorali
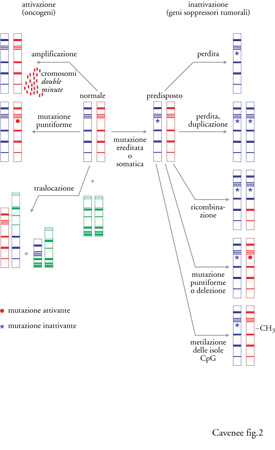
Come menzionato nell'introduzione, nei tumori umani sono stati riscontrati sia la perdita sia l'acquisto di informazioni genetiche. A livello dei geni, la perdita riflette l'inattivazione dei geni soppressori tumorali. La perdita cromosomica è la più comune modificazione genetica osservata nella maggioranza dei tumori umani (Rodriguez et al. 1994), con la sola eccezione della leucemia e del linfoma. Lo smascheramento di alleli tumorali recessivi tramite la perdita della eterozigosità è stato discusso nel contesto dei tumori sporadici ed ereditari. I meccanismi attraverso i quali avviene la LOH sono mostrati nella fig. 2. Esempi di questi meccanismi sono stati riscontrati inizialmente nel retinoblastoma sporadico ed ereditario (Cavenee et al. 1983) e in seguito praticamente in tutti i tipi di cancro umano.
Per alcuni geni soppressori tumorali operano tutti e cinque i meccanismi, sebbene uno di essi possa essere predominante. I primi due meccanismi elencati, perdita cromosomica e perdita/duplicazione, risultano nella LOH per tutti i loci di un particolare cromosoma e nel conseguimento dell'omozigosi per la mutazione iniziale. I tumori del cervello costituiscono un esempio di perdita cromosomica: la monosomia del cromosoma 10 è frequentemente associata alla progressione della forma tumorale più maligna, il glioblastoma. Un esempio di perdita cromosomica, seguita dalla duplicazione del cromosoma mutante, è quello del cromosoma 3 nel melanoma dell'uvea. In contrasto con questi meccanismi, la mutazione seguita da ricombinazione somatica risulta nella LOH esclusivamente per una porzione del cromosoma che comprende la regione contenente il gene mutato. L'identificazione delle regioni cromosomiche nelle quali si verificano le ricombinazioni e le delezioni comuni a molti tumori spesso porta alla localizzazione cromosomica dei geni soppressori tumorali, ed è chiamata 'mappatura per delezione'. Il gene p16/CDKN2 è stato identificato attraverso l'uso estensivo di questa tecnica sul cromosoma 9p21. In molti e diversi tipi di tumore, la delezione omozigote è un meccanismo comune di inattivazione del gene p16/CDKN2 e del gene p19 sovrapposto (Pollock et al. 1996). Le mutazioni puntiformi, principali responsabili dell'inattivazione del gene p53 (Hollstein et al. 1996), vengono identificate attraverso tecniche di genetica molecolare e sequenziamento diretto dei nucleotidi. Come suggerito dalla fig. 2, mutazioni puntiformi separate su entrambi gli alleli potrebbero inattivare completamente la normale funzione del gene soppressore senza LOH, sebbene la frequenza di questo evento sia sconosciuta. La natura e la frequenza delle mutazioni in centinaia o addirittura migliaia di tumori sono state catalogate per diversi geni e sono reperibili in varie pubblicazioni (per es., per il gene p16/CDKN2, Pollock et al. 1996; per il gene Rb, Beroud et al. 1996) o su Internet (BRCA1, breast cancer information consortium, BIC). La catalogazione delle mutazioni ha fornito molteplici evidenze sulla presenza di 'punti caldi' (hot spots) per le mutazioni all'interno dei geni, come i codoni 175, 248 e 237 nel gene p53, che possono rendere conto di quasi un quarto di tutte le mutazioni di questo gene, sia considerando tutti i tipi di tumore (Hollstein et al. 1996) sia solo tipi particolari (per es., il tumore del cervello; Bögler et al. 1995). In molti geni del cancro, punti caldi per le mutazioni spesso indicano domini funzionali critici della proteina codificata; per esempio, quelli del gene p53 riguardano domini proteici essenziali per l'interazione con il DNA.
La metilazione delle isole CpG è forse il meccanismo d'inattivazione dei geni soppressori tumorali meno studiato. Le isole CpG sono brevi tratti di DNA ricchi di nucleotidi CpG e sono presenti all'estremità 5′ di circa il 50-60% di tutti i geni noti. Le isole CpG si mantengono in uno stato demetilato in tutti i tessuti normali ma, con un meccanismo non ancora scoperto, quelle alle estremità 5′ di numerosi geni soppressori tumorali diventano metilate nelle cellule tumorali, parallelamente alla perdita dell'espressione del gene. Tali 'mutazioni' non alterano la sequenza nucleotidica ma influenzano l'espressione genica e sono pertanto considerate epigenetiche. Il cosiddetto 'silenziamento epigenetico anomalo', tramite metilazione delle isole CpG nelle cellule tumorali, è stato descritto per i geni VHL, Rb, p16/CDKN4 ed E-caderina (Jones 1996). Dal momento che molti geni soppressori tumorali vengono inattivati prevalentemente da un solo meccanismo, è possibile che esistano geni soppressori inattivati in prevalenza tramite metilazione delle isole CpG. Questi geni sarebbero sfuggiti alle analisi genetiche convenzionali del DNA dei tumori. Recentemente, proprio un gene candidato soppressore tumorale, HIC-1 (hypermetilated in cancer), è stato identificato analizzando una particolare regione cromosomica che presenta un'alterata metilazione delle isole CpG; si è visto che questo gene è alterato con alta frequenza negli stadi precoci dello sviluppo del tumore cerebrale. I pattern di metilazione aberrante delle isole CpG attraverso il genoma sono non casuali e tumore-specifici, suggerendo la possibilità che questi eventi forniscano un vantaggio per la crescita delle cellule tumorali e che inibendo la trascrizione di specifici sottogruppi di geni contribuiscano alla formazione di ben determinati tipi di tumore (Costello et al. 2000). L'integrazione della metilazione con le analisi genetiche indica che la metilazione aberrante colpisce prevalentemente un diverso gruppo di geni piuttosto che meccanismi genetici (Zardo et al. 2002). Ciò suggerisce che la sola analisi genetica dei meccanismi tumorali è probabilmente incompleta, e che potrebbero esserci ulteriori sottogruppi di geni tumorali ancora da identificare.
Oncogeni
La scoperta di oncogeni dominanti ha effettivamente preceduto di circa dieci anni quella dei geni repressori ed è stata realizzata grazie allo studio dei retrovirus trasformanti (Bishop 1995). L'idea che gli stessi retrovirus potessero essere responsabili del cancro, o che l'identificazione dei geni trasformanti codificati da questi virus potesse condurre all'identificazione di analoghi geni tumorali endogeni nell'uomo, si è rivelata fondata. Un numero limitato di tumori è fortemente associato a specifiche infezioni virali; esso comprende il linfoma di Burkitt (BL) e il carcinoma naso-faringeo associati al virus di Epstein-Barr, il tumore del fegato associato al virus dell'epatite B, il tumore della cervice uterina al papillomavirus e la leucemia delle cellule T al relativo virus (Varmus e Weinberg 1993). Degli oltre venti oncogeni identificati grazie all'omologia con i geni trasformanti dei retrovirus, solo per pochi (KRAS, HRAS, ERBB, ERBB2, MYC, NMYC) è stato dimostrato un coinvolgimento nel cancro umano. Attualmente più di cento geni sono ritenuti oncogeni, sebbene ciò dipenda probabilmente dall'inclusione di tutti i geni con capacità trasformante dominante, a prescindere dall'effettivo coinvolgimento nel cancro umano (Bishop 1995). La comprensione dei meccanismi cromosomici che attivano gli oncogeni ha portato all'identificazione della maggior parte di tali geni, noti per il loro contributo nelle neoplasie umane. I protooncogeni cellulari, geni che normalmente hanno un ruolo positivo nella regolazione della crescita, possono essere modificati in forme oncogeniche mediante amplificazione genica, mutazioni puntiformi o traslocazioni reciproche (fig. 2). L'amplificazione genica è definita come l'incremento numerico (che può essere di oltre cento volte in alcune cellule cancerose) di segmenti di DNA compresi tra 100 kb e 2-3 Mb (Amler e Schwab 1992; Gaudray et al. 1992) e può presentarsi in una delle due seguenti anomalie citogenetiche: come regioni che si colorano in modo omogeneo (HRS, homogeneously staining regions) quando fanno parte di un cromosoma, oppure sotto forma di minicromosomi soprannumerati, detti DM (double minutes), quando sono extracromosomici. Le regioni amplificate, o 'ampliconi', spesso contengono numerosi geni e nella maggior parte dei casi, ma non sempre, vi è una corrispondente sovraespressione del gene amplificato (o dei geni amplificati) che contribuisce all'aumentato potenziale di crescita e al carattere maligno delle cellule affette. Il gene amplificato può anche essere riarrangiato, diventando un oncogene sovraespresso, costitutivamente attivo e deregolato. L'amplificazione genica è un evento che si osserva essenzialmente nelle fasi tardive dello sviluppo del tumore ed è molto probabilmente correlata alla progressione, piuttosto che alla fase iniziale dei tumori (Rodriguez et al. 1994).
L'attivazione degli oncogeni per mezzo di mutazioni puntiformi produce in genere un segnale positivo di crescita che è attivo in modo costitutivo, come nel caso delle mutazioni dei geni RAS e BRAF (Davies et al. 2002). Il gene RAS è importante per la trasduzione dei segnali da vari recettori della superficie cellulare, inclusi quelli per i fattori di crescita (Bos 1995). Coadiuvato da un gruppo di proteine regolatrici e adattatrici, il segnale trasdotto da RAS alla fine raggiunge il nucleo e attiva l'espressione dei geni responsabili dello sviluppo. Mutazioni puntiformi nel gene RAS, presenti nel 25% di tutti i tumori umani e fino al 90% in certi tipi di tumore (per es., nei carcinomi del pancreas), comportano una continua trasduzione del segnale via RAS, anche in assenza di segnali extracellulari. Le mutazioni puntiformi di RAS sono altamente specifiche e quasi tutte riscontrate nei codoni 12, 13 o 61.

Un elenco di oncogeni attivati per amplificazione genica o per mutazioni puntiformi, inclusi quelli precedentemente discussi, è presentato nella Tav. I.D.
Questi oncogeni possono essere suddivisi in cinque gruppi in base alle loro funzioni note e alla localizzazione cellulare e comprendono fattori di crescita extracellulari, recettori transmembrana dei fattori di crescita, trasduttori intracellulari di segnali e fattori nucleari di regolazione del ciclo cellulare e di trascrizione. Indipendentemente dal meccanismo di attivazione, questi oncogeni agiscono in modo dominante per promuovere il carattere maligno.
Un terzo meccanismo per l'attivazione degli oncogeni è la traslocazione cromosomica (fig. 2), che è il meccanismo eziologico primario nella leucemia e nel linfoma (Rabbits 1994). La traslocazione implica due rotture cromosomiche, solitamente su due cromosomi diversi, seguite dallo scambio reciproco e dal ricongiungimento di un segmento di un cromosoma con quello di un altro. Le inversioni cromosomiche sono meno comuni e coinvolgono eventi in un singolo cromosoma, quali la rottura, l'inversione e il riattacco. Le due principali conseguenze delle inversioni e delle traslocazioni sono: (1) la giustapposizione di un gene che codifica per una immunoglobulina o per un recettore genico della cellula T con un protooncogene, così da attivare quest'ultimo e farlo esprimere in maniera aumentata e non regolata; (2) una fusione genica conseguente alla riunificazione, che codifica una proteina trasformante chimerica, nel caso che ambedue i punti di rottura cromosomica siano situati all'interno di introni.

Nella leucemia e nel linfoma, il consistente e frequente coinvolgimento dei geni per le catene pesanti (IgH) e leggera (IgL) delle immunoglobuline e dei geni per i recettori delle cellule T nell'attivazione dei protooncogeni sembra riflettere aberrazioni nel normale processo con cui questi geni vengono riarrangiati nelle cellule ematopoietiche in modo da generare i geni per i recettori degli antigeni. La prima traslocazione, caratterizzata dal punto di vista molecolare, consiste in una giustapposizione del protooncogene Myc, già noto per essere attivato mediante amplificazione genica nei tumori solidi, con un gene IgH in una neoplasia maligna dei linfociti B, il linfoma di Burkitt. La traslocazione di Myc e la conseguente sovraespressione si verificano nel 90% dei casi di BL. La sovraespressione del gene Myc (che codifica un fattore di trascrizione) altera l'equilibrio di una serie di fattori di trascrizione coinvolgendone almeno altri tre, MAD, MAX e Mxi-1. La sovraespressione di Myc sposta l'equilibrio verso un eccesso di dimeri Myc-MAX, necessari per l'attività dell'oncogene. Altri geni con ruoli diversi nella regolazione dello sviluppo (Hox11) nel differenziamento (TAL1/SCL) e nell'apoptosi (BCL-2) sono attivati o sovraespressi da simili eventi di traslocazione (Tav. I.E).


Paragonate all'attivazione dei protooncogeni, la fusione genica e l'espressione di proteine chimeriche potrebbero essere le conseguenze più comuni degli eventi di traslocazione (Tav. Ia e Ib.F).
Queste traslocazioni reciproche generano due potenziali fusioni, le quali potrebbero essere espresse, ma in genere nelle cellule tumorali si ritrova solo un trascritto di fusione. Nella stessa cellula possono anche essere presenti i trascritti dei geni normali di ciascun omologo non coinvolto nella traslocazione, cosicché le proteine chimeriche possono contribuire al carattere maligno mediante un meccanismo dominante negativo. Nelle cellule della leucemia mieloide acuta (AML, acute myelogenous leukemia) sono state dimostrate in vitro l'inibizione dominante negativa del recettore normale dell'acido retinoico (RARa), causata dalla fusione PLZF-RARa, e quella della proteina normale AML (CBFA2, una subunità del fattore di trascrizione CBF) da parte di entrambe le proteine di fusione AML1/MDS1 o AML/EAP. Tuttavia, nel 50% delle leucemie linfocitiche acute con traslocazioni di ALL-1, il gene ALL-1 (chiamato anche MLL o Hrx) si fonde con sé stesso lasciando la cellula affetta senza alcun gene ALL-1 normale, con un fenomeno che ricorda l'inattivazione dei geni soppressori tumorali. Le cellule staminali embrionali (ES, embrionic stem) con un doppio knock out del gene ALL-1 mostrano colonie con due fenotipi, simili alle cellule ematopoietiche anormali dei pazienti leucemici e crescono più velocemente delle cellule parentali o di quelle ES in cui è stato inattivato un solo gene ALL, suggerendo che la perdita di funzione del gene ALL potrebbe contribuire al carattere maligno (Fidanza et al. 1996). Da questi studi emerge l'ipotesi che ALL possa agire come un gene soppressore tumorale piuttosto che come un oncogene dominante.
Le fusioni geniche identificate nelle neoplasie maligne delle cellule ematopoietiche spesso implicano fattori di trascrizione; tuttavia sono state identificate anche fusioni con proteine che legano RNA (TLS/FUS), con domini della tirosinachinasi (ABL), omeodomini (HOX9A) e con le proteine dei pori nucleari (NUP98, CAN/NUP214). Il primo prodotto di fusione caratterizzato molecolarmente è stato la fusione BCR-ABL, presente in quasi tutte le leucemie mielogene croniche (CML) e spesso anche nelle leucemie linfoblastiche acute (ALL). Questo rappresenta anche il primo caso di traslocazione reciproca caratterizzato dal punto di vista citogenetico: il cromosoma 22 risultante è stato denominato 'cromosoma Philadelphia'. Il gene ABL, l'omologo umano dell'oncogene virale che provoca la leucemia murina di Ableson, codifica una tirosinachinasi citoplasmatica, mentre BCR codifica una proteina con una funzione sconosciuta. La conseguenza principale della fusione BCR-ABL è un'aumentata e non regolata attività tirosinochinasica; quindi, si tratta probabilmente di una mutazione del tipo 'acquisto di funzione'. Topi transgenici BCR-ABL e topi normali irradiati, che hanno subito un trapianto di midollo osseo con cellule che esprimono BCR-ABR, sviluppano neoplasie maligne, indicando che BCR-ABL agisce in modo dominante nel provocare lo sviluppo del carattere maligno. Un nuovo inibitore di questa fusione superattiva di tirosinachinasi, il Gleevec, è stato utilizzato per trattare con successo alcuni di questi pazienti leucemici. Il Gleevec ha anche mostrato una notevole attività antitumorale contro i tumori gastrointestinali causati dall'oncogene c-Kit.

Le traslocazioni che generano le proteine di fusione si verificano anche nei tumori solidi. Questi studi sono stati focalizzati sui sarcomi perché in essi le anomalie citogenetiche sono ben caratterizzate. La prima fra queste fusioni geniche a essere studiata riguardava il gene RET nel carcinoma papillare della tiroide (PTC, papillary thyroid carcinoma). Il dominio della tirosinachinasi del gene RET è fuso con uno di tre geni differenti, la cui funzione è ancora sconosciuta e scarsamente definita (Tav. I.G).
Le tre fusioni di RET sono state trovate nel PTC. Il gene del sarcoma di Ewing (EWS) può fondersi con uno di quattro partner di traslocazione, tra cui WT1 (Gerald et al. 1995), il gene soppressore tumorale responsabile di alcuni casi di tumore di Wilms. A differenza delle traslocazioni di RET, ognuna delle quattro traslocazioni cui EWS può andare incontro è stata osservata in un differente tipo di tumore (Tav. I.C). Non è ancora chiaro se ciascuna traslocazione di EWS sia specifica di un tipo particolare di tumore.
La progressione maligna di cellule premaligne
Il modello di evoluzione clonale

Finora abbiamo discusso di mutazioni singole nei tumori; tuttavia, è chiaro già da tempo che lo sviluppo del tumore richiede molte tappe. Il processo attraverso il quale da una singola cellula normale si sviluppa un tumore clinicamente evidente viene definito 'evoluzione clonale' ed è riassunto nella fig. 3. In questo modello genetico (Nowell 1976), lo sviluppo del cancro ha inizio con una mutazione somatica o ereditaria in una singola cellula, ed è seguito da una LOH, in accordo con l''ipotesi dei due eventi' che è stata formulata da Alfred G. Knudson. In questo modello, le prime cellule tumorali sono con molta probabilità normali secondo criteri morfologici e biochimici, ma hanno acquisito un vantaggio proliferativo rispetto a quelle normali adiacenti. Il clone tumorale iniziale continua a dividersi sviluppando cellule tumorali geneticamente identiche. Occasionalmente, la divisione cellulare verrà accompagnata da un'ulteriore mutazione; molte di queste mutazioni saranno letali mentre altre non conferiranno alcun vantaggio selettivo. I rari casi in cui esse conferiscono un vantaggio positivo nella crescita rappresentano le basi molecolari attraverso le quali le cellule tumorali evolvono verso un aumentato carattere maligno. Il numero di queste mutazioni vantaggiose, inizialmente rare, tende ad aumentare con la divisione cellulare, talora abbastanza rapidamente, in concomitanza con un riscontro clinico di un passaggio netto da una crescita benigna a un tumore a carattere fortemente maligno. Nelle popolazioni umane, l'accumulo dei difetti genetici è tempo-dipendente, in accordo con l'aumento dell'incidenza del cancro con l'età.
Le alterazioni genetiche che conferiscono un vantaggio di crescita possono essere di diversa natura. Le mutazioni possono modificare molecole che regolano la crescita normale (per es., geni soppressori tumorali), permettere la crescita di cellule tumorali in modo indipendente dagli ormoni o indurre, mediante effetti locali, cambiamenti circoscritti nella crescita. Cellule con alterazioni genetiche o epigenetiche che conferiscono resistenza a sostanze chimiche o alle radiazioni potrebbero anche essere favorite dalle pressioni selettive che provengono dalle terapie anticancro. In uno stadio qualsiasi della progressione del tumore, differenze nell'ambiente locale possono stimolare l'insorgenza di subcloni varianti, il più 'adatto' dei quali supererà alla fine gli altri, divenendo la popolazione dominante del tumore in evoluzione. Si può anche verificare una divergenza di più subcloni lungo differenti vie di progressione, contribuendo in tal modo all'eterogeneità genetica osservata in alcuni tumori. Perciò, l'evoluzione clonale delle popolazioni di cellule tumorali è un processo a più tappe, trainato dall'acquisizione continua e progressiva di vantaggi genetici di crescita e di modificazioni epigenetiche in un tipo cellulare che una volta possedeva un genotipo e un fenotipo normali.
Evidenze a favore del modello
L'origine monoclonale della maggior parte dei tumori umani è stata verificata con differenti marcatori genetici ed epigenetici. Nelle neoplasie ereditarie, il tumore origina da una cellula eterozigote predisposta in cui si verifica una seconda mutazione nella restante copia normale del gene. Con pochissime eccezioni (gene RET), tutte le cellule dei tumori ereditari sono omozigoti per il gene soppressore tumorale mutante e quindi devono derivare da un'unica o al massimo da poche cellule. La dimostrazione dell'origine unicellulare della maggior parte dei tumori sporadici è stata più ardua, perché non si conosceva quali fossero i difetti iniziali, e nemmeno se essi fossero genetici o epigenetici (o entrambe le possibilità).
Mediante l'analisi citogenetica si è evidenziato per la prima volta che nella leucemia mieloide cronica (CML) il 100% delle cellule tumorali presentava la stessa specifica anomalia dei cromosomi 9 e 22, suggerendo che questa alterazione genetica era avvenuta nella cellula tumorale originaria. I geni interessati, BCR e ABL, sono stati isolati, rispettivamente sul 9 e sul 22, e la presenza della mutazione BCR-ABL è stata dimostrata in tutte le cellule, a riprova di un'origine unicellulare della CML a livello genico. L'origine clonale o unicellulare della CML è stata inoltre dimostrata grazie agli studi sulle modalità di inattivazione del cromosoma X in cellule normali e affette da CML prelevate dallo stesso paziente. Nelle cellule delle femmine, uno dei due cromosomi X viene inattivato in modo casuale ed ereditario. Mentre i tessuti normali sono costituiti da un mosaico di cellule, il tessuto tumorale, se di origine clonale, dovrebbe avere lo stesso cromosoma X inattivo in tutte le cellule. L'espressione dei geni polimorfici del cromosoma X, come quello per la glucosio-6-fosfatodeidrogenasi (G6PD), consente di verificare queste possibilità. Le cellule CML di individui polimorfici esprimono solo un tipo di G6PD, indicando che tutte le cellule CML devono essersi originate per proliferazione di una singola cellula. L'impiego di questo tipo di indagini sull'origine clonale delle cellule tumorali, sfruttando l'inattivazione del cromosoma X, ha permesso di estendere tali conclusioni alla maggioranza dei tumori sporadici comuni.
Con l'analisi di campioni tumorali ottenuti sequenzialmente dallo stesso individuo, o di campioni conseguiti sincronicamente, contenenti componenti distinguibili di differenti stadi della progressione, è stata dimostrata la parallela progressione clinica e genetica dei tumori umani, ipotizzata nel modello di evoluzione clonale. I tumori che richiedono l'asportazione chirurgica, ma che successivamente si ripresentano in forma più maligna nello stesso sito, come i tumori al cervello, permettono l'analisi sequenziale delle alterazioni genetiche. Coppie di tumori cerebrali dello stesso paziente, uno a basso e uno ad alto grado di malignità, sono state esaminate per la mutazione del gene soppressore tumorale p53, in modo tale da permettere una valutazione quantitativa degli alleli p53 mutanti rispetto a quelli normali in ciascun tumore (Sidransky et al. 1992). I tumori a bassa malignità presentavano solamente poche cellule con mutazioni specifiche del gene p53, mentre i tumori ricorrenti altamente maligni erano composti quasi esclusivamente da cellule contenenti il mutante p53. La mutazione specifica (un cambiamento di un singolo nucleotide) era identica in entrambi i tumori di ciascuna coppia; ciò ha portato alla conclusione che una rara cellula mutante, nel tumore a bassa malignità, era andata incontro a un'espansione clonale tale da costituire il tipo cellulare predominante nel tumore ad alta malignità. Un singolo tumore cerebrale, che presentava componenti sia a basso sia ad alto grado di malignità, è stato dissezionato e analizzato in modo analogo. Nella porzione a bassa malignità il 60% delle cellule conteneva un gene p53 mutante e uno normale e il 40% delle cellule aveva due differenti geni p53 mutanti, mentre la componente ad alto grado di malignità era composta interamente da cellule con entrambi gli alleli mutanti. La progressione istologica e clinica dei tumori cerebrali è stata quindi associata all'espansione clonale di cellule che avevano precedentemente acquisito una mutazione nel gene p53 che aveva a sua volta fornito loro un vantaggio selettivo nella crescita.
L'accumulo sequenziale di difetti genetici è stato dimostrato anche in molti altri studi. Per esempio, con l'analisi citogenetica di un neurofibrosarcoma prelevato tre volte nell'arco di cinque mesi sono state osservate mutazioni cumulative. Il primo campione, ottenuto da biopsia con ago aspirato, presentava come unica anomalia clonale citogenetica un sovrannumero di i(1)q10; alcune cellule analizzate avevano perduto anche il cromosoma 18. Tre settimane dopo, un secondo campione, ottenuto per resezione chirurgica, mostrava un cariotipo predominante con i(1)q10, monosomia del cromosoma 18 e trisomia del cromosoma 21. Dopo cinque mesi è stato asportato un grande tumore ricorrente nel cui cariotipo complesso erano presenti alcune anomalie trovate nei primi due campioni, assieme a diverse nuove aberrazioni. La divergenza genetica e l'alto grado di anomalie citogenetiche riscontrato nel corso della progressione di questo tumore si osservano anche in molti altri tipi di neoplasia, e sono particolarmente evidenti nei tumori microdissezionati che mostrano instabilità genomica.
Per studiare i vari aspetti del modello di evoluzione clonale proposto, quando per motivi clinici era difficile disporre di campioni sequenziali, sono stati utilizzati anche campioni ottenuti contemporaneamente dallo stesso tumore, contenenti componenti distinte con differenti gradi di malignità. Il dubbio che i campioni sincroni appartengano a un continuum di progressione maligna è stato chiarito, per quanto riguarda diversi tipi di tumore, come quelli della mammella, del polmone, del colon e dell'uvea, grazie alla dimostrazione di alterazioni genetiche specifiche condivise dai vari campioni. Per esempio, in una metaplasia squamosa (un'anomalia non tumorale dei bronchi) adiacente a un carcinoma a cellule squamose, ma fisicamente separata da esso, era presente una piccola frazione di cellule con una specifica mutazione del p53 identica alla mutazione che si osserva nelle cellule del carcinoma: ciò dimostra chiaramente una discendenza nella progressione maligna. Similmente, la progressione del carcinoma del dotto in situ (DCIS, ductal carcinoma in situ) a tumore della mammella, infiltrato e metastatico, è stata dimostrata grazie all'identificazione, in entrambi i tipi tumorali, di sette regioni cromosomiche che presentavano LOH. Tuttavia, in questi stessi individui, foci (focolai di infezione latente) di DCIS adiacenti mostravano un tipo diverso di perdita allelica negli stessi loci cromosomici, indicando una divergenza genetica nel corso dell'evoluzione clonale di DCIS a tumore invasivo della mammella. In vari pazienti con tumore del colon sono stati osservati contemporaneamente adenoma benigno di grado elevato e carcinoma maligno, ed è stata utilizzata la tecnica di 'tipizzazione allelica' per dimostrare l'avvenuta LOH in siti cromosomici vicini o all'interno di tre geni soppressori tumorali, il gene APC sul cromosoma 5q, il gene DCC sul 18q e il gene p53 sul 17p (Boland et al. 1995). L'espansione clonale di cellule con LOH nel cromosoma 5q si verifica precocemente negli adenomi benigni e quella di cellule con LOH nel cromosoma 17q ha luogo durante la transizione da grandi adenomi a carcinoma. La displasia grave, che precede il carcinoma, era caratterizzata da un alto grado di eterogeneità genetica, mostrando, in questa fase della progressione del tumore, una divergenza. La LOH del cromosoma 18q non è stata associata in modo preferenziale nessuno degli stadi patologici.
Un recente esempio di un melanoma dell'uvea con una componente pigmentata e una non pigmentata ha illustrato in modo drammatico il parallelismo tra la progressione istologica e quella genetica. Le due componenti sono facilmente dissezionabili e patologicamente distinte, con la porzione pigmentata composta da cellule epitelioidi di forma regolare e quella non pigmentata contenente un insieme più eterogeneo di "grandi cellule epitelioidi e molte forme bizzarre e multinucleate". L'analisi citogenetica ha mostrato cinque anomalie nelle cellule pigmentate e almeno undici in quelle tumorali non pigmentate. Tre delle undici alterazioni sono comuni a entrambe le componenti; una quarta anomalia, la monosomia del cromosoma 3, è stata rilevata solo nelle cellule pigmentate con l'analisi citogenetica, ma l'analisi genetica a livello molecolare ha evidenziato LOH nel cromosoma 3 nelle cellule non pigmentate. Ciò indica che la disomia del cromosoma 3 nelle cellule non pigmentate è dovuta a monosomia seguita da duplicazione. Sia l'analisi genetica sia l'istopatologia suggeriscono con forza che le cellule tumorali non pigmentate pleomorfiche derivano dall'espansione clonale e dalla successiva divergenza di cellule pigmentate omogenee.
È molto interessante notare che condizioni descritte come 'non tumorali', quali la metaplasia squamosa del polmone, l'adenoma benigno del colon e la presenza di cellule anormali della mucosa nel cancro della testa e del collo, presentano una frazione di cellule con mutazioni genetiche. È possibile che si tratti, in realtà, di cellule tumorali a uno stadio molto precoce dell'evoluzione del tumore. Queste osservazioni sono di importanza decisiva, poiché mostrano la possibilità di utilizzare marcatori genetici per identificare cellule potenzialmente maligne a uno stadio della loro progressione molto più precoce di quanto non fosse possibile in passato.
Esempi di evoluzione clonale: cooperazione dei geni soppressori tumorali e oncogeni
Una comprensione accurata ed esauriente dell'eziologia genetica del cancro umano necessita dell'integrazione di un insieme di anomalie che coinvolgono sia gli oncogeni sia i geni soppressori tumorali. Per una varietà di tumori, tra cui quelli del cervello, del colon, del polmone, della cervice uterina, della mammella e della prostata, si sta tentando di individuare questo tipo di progressione genetica integrata. La ben definita progressione istopatologica dei tumori del cervello e del colon fornisce una base per classificare queste anomalie genetiche ed è servita da prototipo per analisi analoghe in altri tipi di tumore. Questi due tipi di cancro sono paradigmatici anche per l'analisi della progressione dei tumori che si ottiene con campioni sequenziali, nel caso del tumore del cervello, e per l'analisi della progressione tumorale di campioni ottenuti contemporaneamente, nel caso del cancro del colon, limitata necessariamente alle componenti dissezionabili.
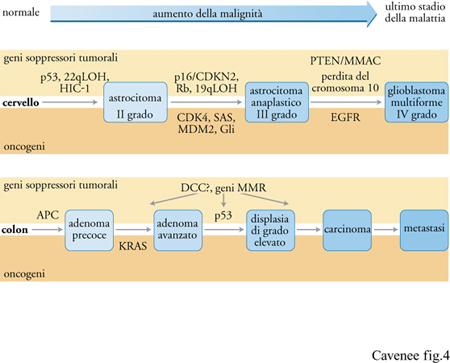
Nella progressione maligna dei gliomi, la forma più comune di tumore cerebrale primario, sono stati distinti tre stadi istopatologici, accompagnati parallelamente da tre serie cumulative di alterazioni genetiche specifiche (fig. 4). Dall'esame di tumori primari e ricorrenti nello stesso paziente e di tumori di vario grado di malignità in pazienti diversi, si evidenzia un accumulo specifico e ordinato di geni soppressori tumorali e di oncogeni. Le alterazioni genetiche che si verificano nelle fasi iniziali della progressione maligna, quali, per esempio, la mutazione di p53, la LOH 22q e la metilazione di HIC-1, sono rintracciabili in tutti gli stadi del glioma (II-IV), mentre eventi genetici tardivi (per es., la perdita di p16/CDKN2 o l'amplificazione di CDK4) si verificano soltanto negli stadi più maligni (III, IV) o quasi esclusivamente (per es., la monosomia del cromosoma 10) nelle malattie in fase terminale (grado IV). Ognuna di queste anomalie si verifica con frequenze alte, ma non sempre, indicando la possibilità di vie alternative che coinvolgano una mutazione o un'altra. Per esempio, la mutazione di p53 si verifica approssimativamente nel 30% di tutti i gliomi (Bögler et al. 1995), l'amplificazione del gene EGFR nel 40% e la monosomia del cromosoma 10 nel 60-95% dei gliomi di IV grado. A differenza dell'alterazione di un singolo gene, la frequenza di inattivazione di una particolare via regolatrice della crescita, che può verificarsi attraverso molteplici tappe, è vicina al 100%. Per esempio, l'abolizione della via di regolazione negativa governata dal gene Rb è il risultato, in quasi tutti i gliomi molto gravi, di uno solo di vari possibili eventi, tra i quali la perdita di Rb, la delezione di p16/CDKN2 o l'amplificazione di CDK4 (e meno frequentemente di CDK6). Similmente, la via di inibizione della crescita cellulare che coinvolge p53 è inattivata, in alcuni gliomi, dalla mutazione di p53 e, in altri, dall'amplificazione/sovraespressione del gene MDM2 e della proteina, da esso codificata, che si lega rendendo inattivo il dominio di attivazione della trascrizione di p53. Potrebbe quindi essere necessaria l'inattivazione di una particolare via regolatrice della crescita per la progressione di alcuni tumori.
Anche la progressione maligna del tumore del colon implica un accumulo di alterazioni negli oncogeni e nei geni soppressori tumorali, dimostrata mediante l'analisi di campioni di tumori di diversi gradi di malignità prelevati contemporaneamente da individui singoli e dal confronto di tumori di molti pazienti (fig. 4) (Fearon 1995; Boland et al. 1995). Numerosi esempi di mutazioni genetiche identiche negli adenomi (stadi precoci) e nei carcinomi (stadi avanzati) dello stesso paziente affetto da tumore del colon convalidano l'uso di campioni ottenuti contemporaneamente nello studio della progressione delle neoplasie maligne. Tre di queste alterazioni genetiche si verificano con alta frequenza e con una specificità legata allo stadio della malattia: la mutazione o la LOH del gene soppressore tumorale APC nel 50% degli adenomi precoci, la mutazione puntiforme del codone 12 o 13 dell'oncogene KRAS nel 50% dei casi di adenomi di grandezza inferiore a 1 cm e la LOH del gene p53 nel 75% dei carcinomi del colon. È anche frequente l'inattivazione del gene DCC, ma non è un evento stadio-specifico.
Nella progressione genetica sia dei tumori del colon sia di quelli del cervello, nonché della maggior parte dei tumori studiati sino a oggi, le alterazioni più precoci riscontrabili riguardano i geni soppressori tumorali, mentre il passaggio a stadi con carattere più maligno è il risultato dell'effetto combinato dell'attivazione di oncogeni e dell'inattivazione del gene soppressore tumorale.
Gli effetti delle mutazioni sul fenotipo
La base genetica del cancro risulta evidente nella consistente associazione tra mutazione, ereditata o acquisita, di geni soppressori tumorali o di oncogeni, con forme o sindromi cancerose specifiche. Nel caso delle sindromi tumorali, un singolo gene mutante può predisporre gli individui a molteplici tipi di tumore, ma l'analisi clinica su più generazioni di famiglie numerose indica che, mentre alcuni tipi di tumore possono essere condivisi dalle famiglie più colpite, l'incidenza di altri tumori varia da una famiglia all'altra. L'indagine sull'origine di tale variabilità, attraverso analisi dettagliate delle mutazioni in molti di questi geni, ha rilevato notevoli correlazioni tra tipi particolari di tumore, osservati in ciascuna famiglia, e tipo e localizzazione della mutazione all'interno del gene responsabile. Descriveremo ora due esempi di queste correlazioni, le mutazioni del gene RET con la neoplasia endocrina multipla di tipo 2 (MEN2) e le mutazioni del gene VHL con il morbo di von Hippel-Lindau.
MEN2 si manifesta con tre varianti clinicamente definite, MEN2A, MEN2B e FMTC, che hanno in comune una predisposizione verso i tumori delle cellule 'C' della tiroide (carcinoma midollare della tiroide o MTC; Mak e Ponder 1996). Queste sindromi differiscono in quanto MTC è l'unico tumore tipicamente presente nelle famiglie FMTC, mentre sia i pazienti MEN2A sia quelli MEN2B possono sviluppare i tumori della ghiandola surrenale (feocromocitomi) e solo i pazienti MEN2A sviluppano iperplasia paratiroidea. Uno spettro molto ristretto di mutazioni del gene RET causa MEN2A, MEN2B e FMTC, e le mutazioni sono specifiche per ognuna di queste sindromi. In MEN2A l'87% delle mutazioni avviene a livello di un singolo residuo di cisteina nel codone 634 e il 93% in una delle cinque cisteine del dominio extracellulare di RET. Inoltre, la manifestazione dell'iperplasia della paratiroide, specifica per MEN2A, è associata a una mutazione da cisteina ad arginina nel codone 634. Più del 90% dei pazienti MEN2B presenta una mutazione da metionina ad arginina nel codone 918, in corrispondenza del sito del dominio tirosinachinasico di RET, deputato al riconoscimento del substrato. Molte delle mutazioni RET in FMTC sono simili a quelle in MEN2A (cinque cisteine) ma sono distribuite in modo più uniforme. Sono state descritte anche alcune famiglie con mutazioni apparentemente specifiche di FMTC nei codoni 768 e 804 di RET.
Sia la collocazione sia il tipo di mutazioni nel gene VHL mostrano forti correlazioni con il fenotipo della malattia VHL. Essa è stata originariamente descritta come una singola entità clinica in cui tutte le famiglie hanno una predisposizione costante agli angiomi della retina e agli emangioblastomi del sistema nervoso centrale, ma anche una predisposizione al carcinoma delle cellule renali (RCC, renal cell carcinoma) e ai feocromocitomi che risulta variabile tra le famiglie VHL. Analizzando più generazioni di una famiglia numerosa, le mutazioni da T a C del nucleotide 505 erano correlate alla totale assenza di RCC, ma alla presenza del feocromocitoma nel 54% degli individui colpiti. Una relazione inversa è stata osservata in una seconda famiglia numerosa con mutazioni da T a C nel nucleotide 686, in cui il 41% degli individui colpiti aveva RCC, ma soltanto l'1% il feocromocitoma. Una terza famiglia con mutazioni da C a T nel nucleotide 712 presentava tutti e quattro i possibili tumori, con frequenze comprese tra il 32 e il 65% degli individui colpiti. Considerazioni sul tipo di mutazione (cambiamento di senso o missense, non-senso o non-sense, microdelezione/inserzione, grande delezione) rispetto al fenotipo della malattia hanno permesso di evidenziare l'esistenza di una correlazione tra mutazioni del tipo cambiamento di senso e famiglie VHS che sviluppavano feocromocitoma, cioè il 96% di queste famiglie aveva mutazioni di cambiamento di senso nel gene VHL.
Correlazioni tra la localizzazione di mutazioni specifiche in un particolare gene e tipi specifici di tumore sono state dimostrate anche per diversi altri geni, incluso il gene p53 nei tumori sporadici (Beroud et al. 1996) e il gene APC in quelli ereditari del colon (Fearon 1995). Questi dati, assieme allo studio dei geni VHL e RET, hanno mostrato in modo evidente il legame inestricabile tra il fenotipo del tumore e i difetti genetici.
In contrasto con gli studi appena descritti, le stesse identiche mutazioni, riscontrate all'interno e tra le famiglie, potrebbero anche essere il risultato di una singola mutazione avvenuta in un progenitore comune, potenzialmente distante (effetto del fondatore), piuttosto che da mutazioni identiche ma indipendenti. La presenza di tali mutazioni fondatrici ancestrali, all'interno di una popolazione umana con origini etniche e geografiche comuni, è stata descritta per MLH1 e MSH2, geni di riparazione del DNA, in diverse famiglie HNPCC (hereditary non polyposis colon cancer) e per il gene BRCA1, nel caso del tumore delle ovaie di tipo familiare. L'identificazione di mutazioni comuni risultanti da un effetto del fondatore o da eventi indipendenti è di immenso valore in quanto permette una determinazione rapida e tecnicamente semplice dei rischi di cancro prima che se ne manifestino i sintomi.
Applicazioni pratiche della genetica del cancro
L'identificazione degli oncogeni e dei geni soppressori tumorali coinvolti nei tumori ereditari e sporadici ha favorito lo sviluppo di un approccio molecolare alla diagnosi, alla prognosi e al trattamento del cancro. Attualmente il risultato più importante apportato da questi studi ai pazienti malati di tumore consiste nella capacità predittiva e nella diagnosi, che in alcuni casi conducono a strategie di intervento più razionali e precoci. È anche possibile che una diagnosi più precoce del tumore, attraverso l'analisi molecolare, possa portare, per alcuni tipi comuni di tumore, a una più alta percentuale di guarigione con i trattamenti attualmente disponibili.
La predisposizione verso la maggior parte dei tumori ereditari discussi in questo saggio può essere determinata grazie all'analisi genetica di individui presintomatici. Queste analisi sono attualmente di routine negli ospedali e per alcuni tipi di tumore sono tecnicamente semplici ed economiche. Per esempio, nel 90% degli individui FAP che hanno ereditato un gene mutante APC esso può essere identificato attraverso uno dei due test molecolari condotti sugli acidi nucleici utilizzando piccoli campioni di sangue (Powell et al. 1993). Questi test identificano le proteine APC mutanti o troncate con una tecnica di traduzione in vitro (saggio di traduzione della proteina), oppure individuano la riduzione allelica endogena della trascrizione del gene APC. In molti casi l'iniziale identificazione di una mutazione APC in un individuo affetto permette poi un test definitivo per gli altri membri della famiglia che non presentano sintomi. Ciò risulta particolarmente utile quando una prima mutazione fondatrice è rappresentata in più generazioni di famiglie numerose, come è stato recentemente scoperto per due famiglie HNPCC con mutazioni nel gene MSH2 o nel gene MLH, e che risultavano identiche in tutti i membri affetti. Una volta identificate, entrambe le mutazioni erano individuabili con una singola reazione PCR. L'analisi presintomatica per le famiglie FAP e HNPCC permette una sorveglianza colonoscopica focalizzata su quegli individui che ereditano il gene mutante, aumentando in questo modo la probabilità di una diagnosi precoce e di una prognosi migliore. Viceversa, per quanto riguarda il tumore del colon, questi test minimizzano la necessità di effettuare ripetuti monitoraggi invasivi in individui non portatori e riducono la preoccupazione di sviluppare un tumore. Simili procedure genetiche, tecnicamente semplici, possono essere effettuate per quei tumori ereditari con uno spettro di mutazioni molto ristretto, come, per esempio, il morbo di von Hippel-Lindau e la neoplasia endocrina di tipo 2. Monitorare le mutazioni in altri geni responsabili di tumori ereditari, quali il gene Rb nel retinoblastoma o il gene BRCA1 nel tumore familiare della mammella, è molto più difficile perché i geni sono molto grandi e le mutazioni sono distribuite in parti estese del gene piuttosto che raggruppate o situate in punti caldi. L'analisi della sequenza del DNA, effettuata con le attuali metodologie, è insufficiente in questi casi, ma lo sviluppo di batterie di oligonucleotidi o 'DNA chips' per un'analisi di sequenza rapida basata sull'ibridazione potrebbe alla fine far superare questa difficoltà.
Sebbene non siano ancora affermati nella pratica medica di routine, sono in corso di sperimentazione metodi non invasivi per la diagnosi precoce di alcuni tumori sporadici. Due di questi metodi misurano le alterazioni del gene p53, che attualmente rappresentano l'alterazione genetica più frequentemente riscontrata in tutti i tumori, come marcatori diretti o indiretti di cellule cancerose. Il primo metodo, il 'clonaggio fagico' dei prodotti PCR del p53 (Sidransky 1995), implica l'analisi diretta del gene p53 mediante amplificazione PCR di piccole quantità di DNA proveniente da cellule dei fluidi corporei. Un 'repertorio' di prodotti PCR del gene p53 viene allestito all'interno di un batteriofago e poi selezionato per la presenza di geni p53 mutanti, mediante ibridazione con sonde oligonucleotidiche normali e mutate. Questo metodo può identificare un unico gene p53 mutante in un contesto di 10.000 cellule normali. Le cellule con p53 mutante sono state individuate nell'urina di pazienti con tumore della vescica, nei campioni di feci prelevati da pazienti affetti da tumore del colon, dalla saliva di pazienti con tumore della testa e del collo e dall'espettorato di pazienti con tumore del polmone. Cellule con p53 mutante sono state effettivamente individuate nei campioni di espettorato analizzato un anno prima che venisse diagnosticato il tumore del polmone, dimostrando così i potenziali benefici clinici di questo approccio molecolare per una diagnosi precoce del tumore.
Il secondo approccio indiretto rileva la presenza di anticorpi diretti contro la proteina p53 con il saggio ELISA (enzyme linked immuno sorbent assay, saggio basato sull'assorbimento del complesso immunologico a cui è legato un enzima; Lubin et al. 1995). Gli anticorpi diretti contro la proteina p53 non sono normalmente presenti nel siero ma, in seguito a una mutazione, la proteina p53 può divenire più stabile e accumularsi nelle cellule tumorali causando, in una piccola percentuale di casi, la produzione di autoanticorpi p53. Il valore di questo test diagnostico, potenzialmente di routine, è stato dimostrato dalla scoperta di anticorpi diretti contro la proteina p53 (e di conseguenza di un'alterazione di p53) nel siero di due fumatori accaniti, molti mesi prima che fosse loro diagnosticato il tumore del polmone. È importante notare che il livello degli anticorpi diretti contro la proteina p53 diminuiva parallelamente alla risposta al trattamento clinico e radiologico. In un altro caso la remissione della malattia in seguito al trattamento era associata a un concomitante ritorno di aumentati livelli di anticorpi diretti contro la proteina p53, indicando che questo saggio può essere utile per un monitoraggio continuo della risposta dei pazienti al trattamento. Uno studio recente multicentrico ha convalidato questo concetto mostrando una correlazione tra la risposta del paziente, la presenza e i livelli di anticorpi diretti contro la proteina p53 nel siero (Lubin et al. 1995). Si è cominciato a utilizzare questo tipo di indagine anche per pazienti affetti da tumore del seno e del pancreas. L'adattamento dei metodi ELISA e di clonaggio fagico per l'individuazione di altre mutazioni genetiche potrebbe ampliare la loro utilità.
Il tentativo di correlare la sopravvivenza dei pazienti e la risposta ai trattamenti con la molteplicità di alterazioni genetiche riproducibili e specifiche nei tumori sporadici è attualmente a buon punto e potrebbe condurre a una migliore stratificazione dei pazienti (ossia a una migliore rappresentatività della popolazione del campione stesso) e a una più appropriata applicazione delle terapie. Per esempio, l'amplificazione del gene EGFR nei tumori del cervello viene associata a una diminuzione del tempo di ricaduta. Questi studi non hanno ancora considerato la possibilità di un'ulteriore stratificazione su base genetica nel 20% dei tumori del cervello con amplificazione e riarrangiamento di EGFR, che nei sistemi sperimentali conferisce un pronunciato aumento del potere tumorigeno. Similmente, sono state anche stabilite correlazioni dirette e quantitative fra alterazioni dell'espressione della ciclina E e l'aggressività del tumore della mammella. L'uso clinico di queste osservazioni genetiche è attualmente limitato a un ruolo ausiliario nell'istologia del tumore. Uno degli scopi dell'analisi genetica dei tumori sporadici è dunque quello di identificare e di trattare in modo più aggressivo i pazienti che presentano una maggiore probabilità di rispondere a terapie specifiche e di sottoporre a trattamenti differenti quelli che, potenzialmente, non rispondono.
La soppressione della crescita delle cellule tumorali maligne è stata realizzata in sistemi sperimentali tramite manipolazione genetica, come, per esempio, la sostituzione dei geni soppressori tumorali, l'inibizione di oncogeni attivati, l'interferenza con processi di angiogenesi e metastasi e l'aumento dell'immunogenicità delle cellule tumorali. Per quanto riguarda il cancro umano, la terapia genica deve superare numerosi ostacoli, come, per esempio, un metodo selettivo ed efficace per l'introduzione di geni nelle sole cellule tumorali, prima che si possa mantenere la promessa di realizzare terapie migliori mediante la medicina molecolare. I recenti successi nello sviluppo di metodi per indirizzare molecole e cellule verso organi specifici, mediante peptidi selezionati in vivo, suggeriscono che questi ostacoli saranno superati nel prossimo futuro (Pasqualini e Ruoslahti 1996).
Bibliografia
Amler, Schwab 1992: Amler, Lucas C. - Schwab, Manfred, Multiple amplicons of discrete sizes encompassing N-myc in neuroblastoma cells evolve through differential recombination from a large precursor DNA, "Oncogene", 7, 1992, pp. 807-809.
Beroud 1996: Beroud, Christophe - Verdier, Frédérique - Soussi, Thierry, p53 gene mutation: software and database, "Nucleic acids research", 24, 1996, pp. 147-150.
Bishop 1995: Bishop, John Michael, Cancer: the rise of the genetic paradigm, "Genes and development", 9, 1995, pp. 1309-1315.
Bögler 1995: Bögler, Oliver - Huang, H.-J. Su - Kleihues, Paul - Cavenee, Webster K., The p53 gene and its role in human brain tumors, "Glia", 15, 1995, pp. 308-327.
Boland 1995: Boland, C. Richard - Sato, Junko - Appelman, Henry D. - Bresalier, Robert S. - Feinberg, Andrew P., Microallelotyping defines the sequence and tempo of allelic losses at tumour suppressor gene loci during colorectal cancer progression, "Nature medicine", 1, 1995, pp. 902-909.
Bos 1995: Bos, Johannes L., p21 ras: an oncoprotein functioning in growth factor-induced signal transduction,"European journal of cancer", 31A, 1995, pp. 1051-1054.
Cairns 1975: Cairns, John, Mutation selection and the natural history of cancer, "Nature", 255, 1975, pp. 197-200.
Cavenee 1983: Cavenee, Webster K. e altri, Expression of recessive alleles by chromosomal mechanisms in retinoblastoma, "Nature", 305, 1983, pp. 779-784.
Cavenee, White 1995: Cavenee, Webster K. - White, Raymond L., The genetic bases of cancer, "Scientific American", 272, 1995, pp. 72-79.
Costello 2000: Costello, Joseph F. e altri, Aberrant CpG island methylation has non-random and tumor type-specific patterns, "Nature genetics", 25, 2000, pp. 132-138.
Davies 2002: Davies, Helen e altri, Mutations of the BRAF gene in human cancer, "Nature", 417, 2002, pp. 949-954.
Fearon 1990: Fearon, Eric R. - Cho, Kathleen R. - Nigro, Janice M. - Kern, Scott E., Identification of a chromosome 18q gene that is altered in colorectal cancers, "Science", 247, 1990, pp. 49-56.
Fearon 1995: Fearon, Eric R., Molecular genetics of colorectal cancer, "Annals of New York Academy of Science", 768, 1995, pp. 101-110.
Fidanza 1996: Fidanza, Vincenzo e altri, Double knock-out of the ALL-1 gene blocks hematopoietic differentiation in vitro, "Cancer research", 56, 1996, pp. 1179-1183.
Gaudray 1992: Gaudray, Patrick - Szepetowski, Pierre - Escot, Chantal - Birnbaum, Daniel - Theillet, Charles, DNA amplification at 11q13 in human cancer: from complexity to perplexity, "Mutation research", 276, 1992, pp. 317-328.
Gerald 1995: Gerald, W. L. - Rosai, J. - Ladanyi, M., Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplasic small round cell tumor, "Proceedings of the National Academy of Science USA", 92, 1995, pp. 1028-1032.
Hahn, Weinberg 2002: Hahn, William C. - Weinberg, Robert A., Modelling the molecular circuitry of cancer, "National review of cancer", 2, 2002, pp. 331-341.
Hollstein 1996: Hollstein, Monica e altri, Somatic point mutations in the p53 gene of human tumors and cell lines, "Nucleic acids research", 24, 1996, pp. 141-146.
Hunter, Pines 1994: Hunter, Tony - Pines, Jonathan, Cyclins and cancer II: cyclin D and CDK inhibitors come of age, "Cell", 79, 1994, pp. 573-582.
Johnson 1996: Johnson, Ronald L. e altri, Human homolog of patched, a candidate gene for the basal cell nevus syndrome, "Science", 272, 1996, pp. 1668-1671.
Jones 1996: Jones, Peter A., DNA methylation errors and cancer, "Cancer research", 56, 1996, pp. 2463-2467.
Knudson 1971: Knudson, Alfred G., Mutation and cancer: statistical study of retinoblastoma, "Proceedings of the National Academy of Science", 68, 1971, pp. 820-823.
Knudson 1993: Knudson, Alfred G., Antioncogenes and human cancer, "Proceedings of the National Academy of Science", 90, 1993, pp. 10914-10921.
Lohmann 1996: Lohmann, Dietrar R. - Brandt, Birgit - Höpping,Wolfgang - Passarge, Eberhard - Horsthemke, Bernard, Spectrum of RB1 germ-line mutations in hereditary retinoblastoma, "American journal of human genetics", 58, 1996, pp. 940-949.
Lubin 1995: Lubin, Richard e altri, Serum p53 antibodies as early markers of lung cancer, "Nature medicine", 1, 1995, pp. 701-702.
Mak, Ponder 1996: Mak, Yin F. - Ponder, Bruce A.J., RET oncogene, "Current opinion in genetics & development", 6, 1996, pp. 82-86.
Miki 1994: Miki, Yoshio e altri, A strong candidate for the breast and ovarian cancer susceptibility gene BRCA 1, "Science", 266, 1994, pp. 66-71.
Moffett 1995: Moffett, Peter e altri, Antagonism of WT1 activity by protein self-association, "Proceedings of the National Academy of Science USA", 92, 1995, pp. 11105-11109.
Mulvihill 1977: Mulvihill, John J., Genetic repertory of human neoplasia, in: Genetics of human cancer, edited by John J. Mulvihill, Robert Warwick Miller, J.J. Fraumeni, New York, Raven Press, 1997, pp. 137-143.
Nowell 1976: Nowell, Peter C., The clonal evolution of tumor cell populations, "Science", 194, 1976, pp. 23-28.
Pasqualini, Ruoslahti 1996: Pasqualini, Renata - Ruoslahti, Erkki, Organ targeting in vivo using phage display peptide libraries, "Nature", 380, 1996, pp. 364-366.
Pollock 1996: Pollock, Pamela M. - Pearson, John V. - Hayward, Nicholas K., Compilation of somatic mutations of the CDKN2 gene in human cancers: non-random distribution of base substitutions, "Genes, chromosomes & cancer", 15, 1996, pp. 77-88.
Ponder 1995: Genetics and cancer: a second look, edited by Bruce A.J. Ponder, Webster K. Cavenee, Ellen Solomon, Cold Spring Harbor (N.Y.), Cold Spring Harbor Laboratory Press, 1995, pp. 219-232.
Powell 1993: Powell, Steven M. e altri, Molecular diagnosis of familial adenomatous polyposis, "The New England journal of medicine", 329, 1993, pp. 1982-1987.
Rabbits 1994: Rabbits, Terence H., Chromosomal translocations in human cancer, "Nature", 372, 1994, pp. 143-149.
Rodriguez 1994: Rodriguez, Enrique - Sreekantaiah, Chandrika - Chaganti, Raju S., Genetic changes in epithelial solid neoplasia, "Cancer research", 54, 1994, pp. 3398-3406.
Sidransky 1992: Sidransky, David e altri, Clonal expansion of p53 mutant cells is associated with brain tumour progression, "Nature", 355, 1992, pp. 846-847.
Sidransky 1995: Sidransky, David, Screening for clonal genetic alterations, "European journal of cancer", 31A, 1995 pp. 1127-1129.
Taylor 2002: Taylor, Michael D. e altri, Mutations in SUFU predispose to medulloblastoma, "Nature genetics", 31, 2002, pp. 306-310.
Varmus, Weinberg 1993: Varmus, Harold E. - Weinberg, Robert A., Genes and the biology of cancer, New York, Scientific American Library, 1993.
Zardo 2002: Zardo, Giuseppe e altri, Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors, "Nature genetics", 32, 2002, pp. 453-458.