
La cromatina e il controllo dell'espressione genica
La cromatina e il controllo dell'espressione genica
Negli eucarioti, il DNA è organizzato nel nucleo in una struttura nucleoproteica nota come 'cromatina'. Per lungo tempo la cromatina è stata considerata un semplice sistema di 'impacchettamento', deputato a immagazzinare i quasi 2 m di DNA genomico umano nel ristretto spazio nucleare, caratterizzato da un diametro di circa 10 μm. Solo recentemente la nostra percezione della funzione della cromatina è cambiata, con l'attribuzione di molteplici funzioni, modulate nel tempo e nello spazio. È infatti noto come la cromatina, già dai suoi primi livelli strutturali rappresenti un ostacolo per il legame alla doppia elica dei fattori trascrizionali e dell'apparato generale della trascrizione, determinando, di conseguenza, una generale inibizione dell'espressione genica. L'attivazione trascrizionale è, quindi, generalmente preceduta o accompagnata da un locale rimodellamento della struttura della cromatina e da cambiamenti dinamici nelle modalità di condensazione del DNA templato.
Nelle cellule eucariotiche queste modificazioni strutturali sono mediate da una complessa serie di attività enzimatiche che, modificando post-traduzionalmente le proteine che compongono la cromatina o alterandone l'associazione con il DNA mediante l'idrolisi di ATP, partecipano attivamente alla regolazione trascrizionale. Infine, le proprietà locali della fibra cromatinica possono essere modulate attraverso l'introduzione o l'eliminazione di specifiche proteine strutturali capaci di influenzarne la dinamicità. La cromatina costituisce, quindi, una piattaforma su cui convergono molteplici segnali biologici che, una volta integrati, determinano risposte molecolari essenziali per una corretta regolazione non solo della trascrizione ma anche di tutti quei processi metabolici che coinvolgono la doppia elica, quali la replicazione, la riparazione e la ricombinazione. In accordo con ciò, molteplici processi cellulari, come la proliferazione e il differenziamento, vengono influenzati da variazioni della struttura cromatinica. È infatti in aumento il numero di patologie umane, di tipo sia neoplastico che sindromico, la cui causa molecolare accertata risiede in mutazioni dei geni che codificano per proteine appartenenti, appunto, ai complessi di rimodellamento della cromatina.
Sarà perciò estremamente importante comprendere quali geni risultino selettivamente deregolati nella cancerogenesi. Questi studi permetteranno lo sviluppo di nuovi e mirati approcci terapeutici (terapia epigenetica). A tale proposito vale comunque la pena ricordare che attualmente sono in uso alcuni inibitori delle istondeacetilasi e delle DNA-metiltransferasi, che sembrano ben tollerati e presentano un'attività clinica efficace contro diversi tipi di neoplasie. È importante sottolineare come un approccio terapeutico di tipo epigenetico sembra capace di influenzare in modo additivo o sinergico l'efficacia di altri trattamenti, quali una chemioterapia più tradizionale o la radioterapia, sebbene il meccanismo molecolare resti ancora ignoto.
Struttura della cromatina

L'unità di base della cromatina è il nucleosoma, costituito da 146 paia di basi (bp) di DNA che si avvolgono per circa due volte intorno a un ottamero di piccole proteine altamente basiche (cariche positivamente), note come 'istoni'. Ogni nucleosoma contiene due molecole di ognuno dei quattro istoni del core (H2A, H2B, H3 e H4), che si organizzano in un tetramero centrale formato da H3 e H4, fiancheggiato da due eterodimeri di H2A e H2B. I nucleosomi, regolarmente spaziati lungo il filamento di DNA, sono separati tra loro da 10÷100 bp di DNA linker (di connessione). Questa configurazione, nota anche come 'fibra da 10 nm' o 'collana di perle', rappresenta il primo livello di organizzazione della cromatina (fig. 2). Ogni istone del core contiene due distinti domini funzionali: un motivo globulare, C-terminale, altamente conservato, detto histone fold, e un'estremità (coda) N-terminale, unica per ogni specie istonica. Il primo dominio, deputato all'assemblaggio istonico, è implicato sia nella eterodimerizzazione specifica con un altro istone (H2A con H2B, H3 con H4) sia nell'avvolgimento del DNA nucleosomiale. Al contrario, le code N-terminali, apparentemente non strutturate, protrudono all'esterno del nucleosoma e non contribuiscono significativamente alla sua formazione, né alla sua stabilità; sono invece implicate nell'organizzazione delle strutture cromatiniche di ordine superiore mediante l'interazione con altre proteine e nucleosomi (fig. 2). Esperimenti in vitro dimostrano, infatti, che la rimozione delle code istoniche permette la formazione della fibra da 10 nm, ma questa struttura non riesce a organizzarsi nei successivi livelli strutturali.
Oltre agli istoni del core, i nucleosomi degli organismi pluricellulari contengono anche una molecola di istone linker, quale H1, che permette l'avvolgimento di altre 20÷30 bp di DNA. Benché la condensazione sia una proprietà intrinseca alla fibra nucleosomiale, la presenza di H1 favorisce la formazione del livello strutturale immediatamente superiore, costituito dalla fibra da 30 nm (fig. 3). Anche i diversi membri della famiglia degli istoni di connessione, pur non derivando evolutivamente dagli istoni del core, sono caratterizzati da un dominio globulare fiancheggiato da due estremità non strutturate: una breve coda N-terminale e una C-terminale, altamente basica e di circa 100 residui amminoacidici. Studi strutturali hanno permesso di dimostrare come il dominio globulare sia essenziale per il legame dell'istone linker al nucleosoma; in particolare, esso interagisce con il DNA avvolto intorno all'ottamero istonico e non con le proteine del core. Al contrario, la coda C-terminale sembra giocare un ruolo fondamentale nella formazione delle strutture cromatiniche di ordine superiore. Come precedentemente accennato, esistono molteplici versioni della proteina H1 e queste sono in genere presenti nella maggior parte delle nostre cellule. Esperimenti di genetica condotti su modelli murini hanno permesso di dimostrare come questi diversi sottotipi sono funzionalmente ridondanti e una sostanziale riduzione dei livelli totali di istoni di connessione porta alla morte dei topi in utero. Se ne deduce che una concentrazione critica di H1 è cruciale per la progressione dello sviluppo embrionale nei Mammiferi.
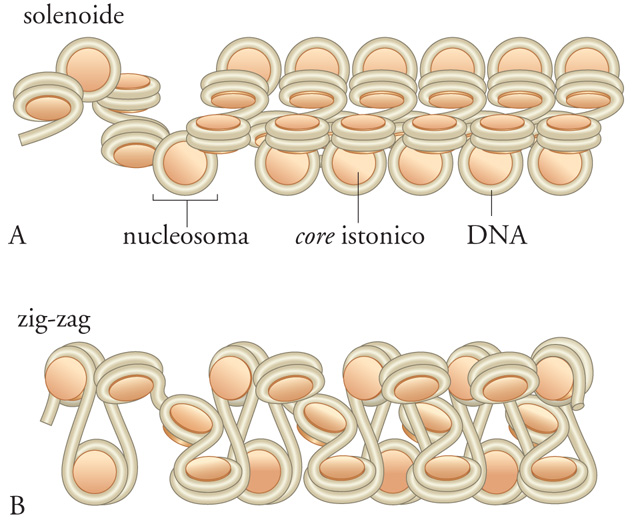
Nonostante oltre venti anni di assidui studi, non si è ancora compreso quale sia la reale struttura della cromatina condensata. L'organizzazione spaziale della fibra da 30 nm, per esempio, viene spiegata da due diversi modelli (fig. 3). Un primo modello propone una struttura solenoidale, in cui 6 nucleosomi consecutivi formano un giro di un'elica che si superavvolge in una spira con un passo di 11 nm. Questa struttura sarebbe determinata e tenuta insieme dalle diverse interazioni tra gli istoni. Un secondo modello, più recente, favorisce una struttura a zig-zag, in cui il legame di H1, producendo un definito angolo di entrata e di uscita del DNA dal nucleosoma, genererebbe una disposizione alternata (a zig-zag) dei nucleosomi. Nonostante le sostanziali differenze fra le due architetture, è molto difficile ottenere evidenze sperimentali che supportino in maniera univoca l'uno o l'altro modello; le più recenti evidenze sembrano comunque favorire quello a zig-zag.
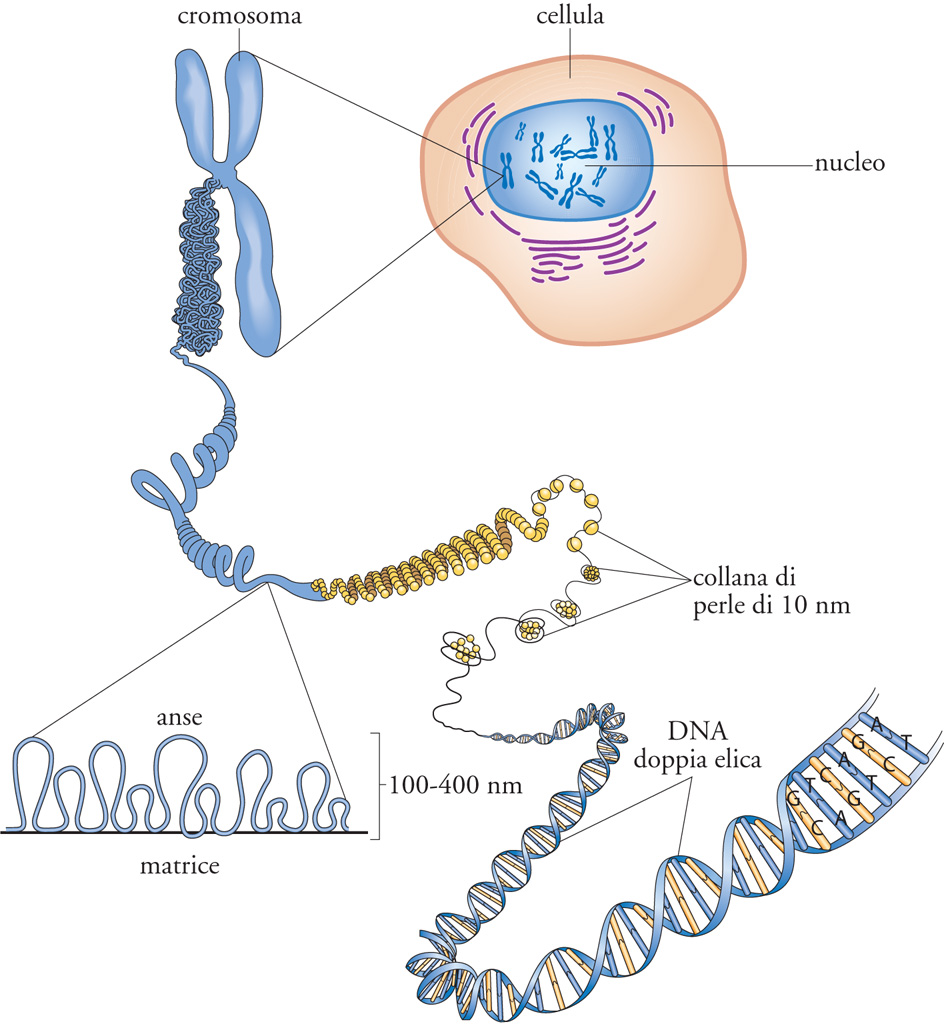
La compattazione del DNA raggiunta con la fibra da 30 nm è ancora insufficiente per alloggiare l'intero genoma nel nucleo della cellula; intervengono, quindi, altri meccanismi di condensazione che determinano la formazione delle fibre dello spessore di 100÷400 nm, che caratterizzano la cromatina interfasica, o strutture ancora più compatte tipiche del cromosoma metafasico. Sebbene la natura di queste strutture non sia ancora nota, diverse prove sperimentali suggeriscono che la fibra da 30 nm formi delle anse di dimensioni variabili da alcune decine di chilobasi fino a più di 10 megabasi. Queste anse sarebbero poi bloccate alla loro base da un complesso proteico denominato scaffold nucleare (fig. 4). Qualunque sia l'organizzazione del DNA all'interno della fibra cromatinica, è oggi ben chiaro che essa ostacola il legame di proteine che devono 'leggere' e/o copiare la sequenza nucleotidica, impedendo di conseguenza la trascrizione. Per questo motivo, negli eucarioti, si è evoluta e affermata una complessa serie di proteine capaci di alterare la struttura della cromatina e di regolarne il livello di compattazione. In particolare, queste funzioni vengono svolte da complessi enzimatici che modificano post-traduzionalmente gli istoni o che, mediante l'idrolisi di ATP, alterano la posizione o la struttura dei nucleosomi. Un terzo meccanismo usato per modulare la conformazione e composizione della cromatina consiste nella sostituzione degli istoni del core con varianti dotate di specifiche caratteristiche strutturali e funzionali.
I complessi di rimodellamento della cromatina ATP-dipendenti
Il processo di trascrizione ha inizio solo se il DNA presente sui promotori è accessibile all'RNA-polimerasi, l'enzima deputato a copiare lo stampo, e ai fattori trascrizionali che ne riconoscono specifiche sequenze; inoltre la doppia elica si deve aprire (denaturare) al passaggio dell'enzima, per richiudersi (rinaturarsi) subito dopo. La struttura della cromatina, già a partire dalla fibra da 10 nm, ostacola tutti questi passaggi indispensabili all'espressione genica, rendendo necessaria la formazione di una struttura più facilmente raggiungibile. I complessi di rimodellamento della cromatina ATP-dipendenti rappresentano una famiglia di proteine che, utilizzando l'energia ottenuta dall'idrolisi dell'ATP e agendo a livello dei nucleosomi, perturbano fisicamente la struttura della cromatina con meccanismi che si sono conservati lungo la scala evolutiva dal lievito fino all'uomo.
Nelle nostre cellule esistono diversi tipi di complessi di rimodellamento ATP-dipendenti che vengono suddivisi in tre famiglie principali, in funzione del tipo di subunità ATPasica; inoltre, all'interno della stessa famiglia l'associazione di questa subunità con diverse proteine porta alla formazione di complessi multiproteici distinti. Inizialmente identificati con analisi genetiche, questi complessi, e in particolar modo quelli appartenenti alla famiglia SWI/SNF e ISWI, sono stati poi largamente studiati in vitro per comprendere le conseguenze del rimodellamento. In vitro possono portare a diversi cambiamenti strutturali, tra i quali possiamo ricordare la distruzione di alcuni contatti tra il DNA e gli istoni all'interno del nucleosoma, lo spostamento di ottameri istonici sia su sequenze fiancheggianti (movimenti traslazionali) sia su molecole di DNA distinte (movimenti in cis e in trans), e un'aumentata accessibilità del DNA nucleosomiale ai fattori di trascrizione. In vivo questi enzimi facilitano lo spostamento dei nucleosomi, la perturbazione dei livelli superiori di organizzazione della cromatina e, molto spesso, l'attivazione trascrizionale di specifici geni.
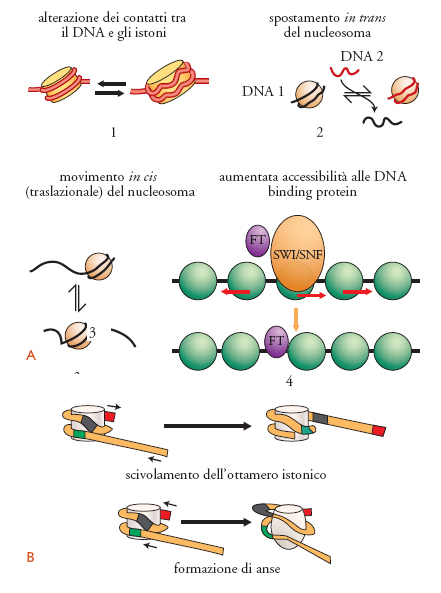
Come fanno questi complessi a catalizzare eventi così diversi? I primi modelli assumevano che il rimodellamento della cromatina avvenisse attraverso la rimozione di istoni appartenenti al nucleosoma. Per esempio, era stato ipotizzato che i complessi della famiglia SWI/SNF utilizzassero l'energia ottenuta dall'idrolisi dell'ATP per rimuovere uno o entrambi i dimeri di H2A-H2B. Recenti studi hanno reso improbabile questa teoria; infatti, il rimodellamento avviene anche su nucleosomi in cui gli istoni sono stati tra loro legati covalentemente. Attualmente i due modelli più accreditati ipotizzano che l'attività di queste proteine si svolga o attraverso un meccanismo di scivolamento dell'intero ottamero istonico, o attraverso la formazione di anse (looping mechanism) sul DNA nucleosomiale (fig. 5). Secondo il primo meccanismo, l'ottamero istonico si sposta su sequenze nucleotidiche fiancheggianti. In questo modello non si ha un aumento della quantità di DNA accessibile, ma solo un cambiamento delle sequenze esposte; inoltre, esso non può giustificare il rimodellamento di sequenze di DNA in cui i nucleosomi sono strettamente impaccati uno vicino all'altro e, quindi, impossibilitati a scivolare. Nel modello ad anse, brevi segmenti di DNA nucleosomiale vengono localmente dissociati dal nucleosoma; in questa fase transitoria, prima di riavvolgersi attorno all'ottamero istonico, il DNA è accessibile ai fattori trascrizionali.
Dal momento che la capacità di rimodellare la cromatina è propria delle subunità centrali contenenti il dominio ATPasico, quale ruolo svolgono le altre proteine appartenenti a questi complessi multiproteici? Attualmente si pensa che queste possano avere una duplice funzione: alcune di esse potrebbero regolare l'attività della subunità catalitica, influenzandone l'efficienza, il risultato finale del rimodellamento o la specificità del substrato, mentre altre potrebbero essere deputate al reclutamento dei complessi su specifici promotori. A questo proposito vale la pena ricordare che in vivo ciascun promotore è caratterizzato da una specifica organizzazione nucleosomiale; è quindi facile ipotizzare che promotori contraddistinti da nucleosomi fortemente impaccati necessitino di complessi capaci di rendere accessibile il DNA attraverso un meccanismo che non richiede lo scivolamento dei nucleosomi. Al contrario, la capacità di spostare traslazionalmente i nucleosomi sarebbe utile su quei promotori contraddistinti da una bassa densità nucleosomiale. Per quanto riguarda la specificità di reclutamento, diverse evidenze sperimentali fanno presupporre che questa sia mediata dalla capacità di alcuni fattori trascrizionali sequenza-specifici di legare direttamente, e quindi portare sul promotore, un ben definito complesso di rimodellamento. In altre parole, ogni gene avrebbe 'scelto' le proprie modalità di regolazione, in accordo con recenti evidenze biochimiche che testimoniano come ogni sistema richieda, per l'attivazione, un solo specifico complesso di rimodellamento. Infine, data la generale capacità di questi enzimi di cambiare la struttura della cromatina, non si può dimenticare come alcuni di essi non siano deputati all'attivazione trascrizionale ma svolgano anche altre funzioni, quali la repressione dell'espressione genica, l'assemblaggio dei nucleosomi o la ricombinazione e la riparazione del DNA.
Le modificazioni degli istoni generano un 'codice istonico'
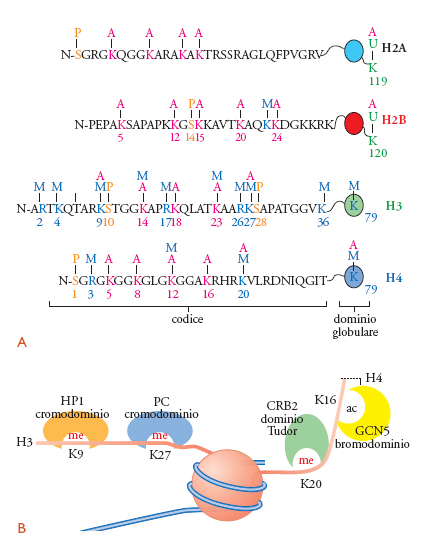
La riorganizzazione della posizione dei nucleosomi da parte di complessi di rimodellamento ATP-dipendenti e la modificazione covalente delle proteine istoniche rappresentano due meccanismi centrali nella regolazione dell'attività cromatinica. In accordo con ciò, le cellule eucariotiche sono dotate di un numero straordinariamente elevato di complessi, capaci di introdurre sulle proteine del nucleosoma una vasta serie di cambiamenti post-traduzionali. L'importanza di questo fenomeno è facilmente apprezzabile se si considera: (a) che prevalentemente le code N-terminali, ma anche alcuni residui del dominio globulare purché situati in una posizione accessibile, possono essere acetilati sulle lisine (K), metilati sulle lisine e le arginine (R), fosforilati sulle serine (S) e le treonine (T), ubiquitinati o sumoilati sulle lisine o ADP ribosilati; (b) che l'espressione genica è influenzata da queste modificazioni e, più in particolare, che in funzione del tipo di modificazione chimica così come del residuo amminoacidico coinvolto si può avere una risposta di tipo attivatorio o repressorio (fig. 6).
Più di quaranta anni fa era stato proposto che l'iperacetilazione dell'estremità N-terminale degli istoni del core fosse coinvolta nell'attivazione dell'espressione genica. L'ipotesi venne rafforzata dalla dimostrazione che in vitro l'acetilazione degli istoni determina un aumento dell'accessibilità del DNA agli enzimi di restrizione e ai fattori trascrizionali. Questo fenomeno veniva spiegato assumendo che tale modificazione, diminuendo la carica netta positiva delle code, ne rendesse più debole l'interazione con il DNA. Il modello fu confermato quando, nel 1996, venne purificata dal protozoo Tetrahymena la prima istonacetiltransferasi (HAT); la proteina isolata, infatti, era l'omologo del fattore GCN5, un ben noto coattivatore trascrizionale di lievito (una proteina che non lega direttamente il DNA, ma lo fa tramite un altro fattore proteico). Il progredire delle ricerche dimostrò non solo che molti altri coattivatori trascrizionali possedevano un'intrinseca capacità di acetilare le lisine degli istoni, ma anche che diversi co-repressori quali, per esempio, Rpd3, erano dotati di un'attività enzimatica opposta, cioè quella costituita dalla capacità di deacetilare gli istoni.
Oggi sappiamo che l'acetilazione è un processo reversibile catalizzato da due gruppi di enzimi. Le istonacetiltransferasi trasferiscono dal cofattore acetil-CoA un gruppo acetilico sul gruppo amminico ε delle lisine degli istoni; al contrario, la rimozione del gruppo acetilico è catalizzata dalle istondeacetilasi (HDAC). La maggior parte delle HAT fa parte di larghi complessi multiproteici che, in analogia con i complessi di rimodellamento ATP-dipendenti, vengono portati specificatamente sui promotori tramite l'interazione con gli attivatori trascrizionali. Analizzando la somiglianza di sequenza, è stato possibile suddividere le HAT in tre famiglie principali, ciascuna caratterizzata da un distinto pattern d'interazione con proteine non istoniche. In vivo, queste formano complessi composti da subunità diverse e, soprattutto, da una differente specificità istonica. L'analisi di topi transgenici privi di una specifica HAT (per es., p300 o PCAF o GCN5) ha chiaramente dimostrato come la singola eliminazione di diverse istonacetiltransferasi possa portare a diversi difetti di sviluppo, confermando come ciascun complesso sia implicato in una specifica funzione biologica. Anche le istondeacetilasi fanno parte di larghi complessi multiproteici, che in questo caso però sono coinvolti nella repressione trascrizionale. Esistono tre principali famiglie di HDAC che differiscono tra loro per dimensione, distribuzione tissutale e meccanismo d'azione; così come per i complessi HAT, anche la diversa composizione di quelli ad attività istondeacetilasica suggerisce che essi siano dotati di distinte funzioni biologiche.
La metilazione è un'altra modificazione post-traduzionale che coinvolge un atomo di azoto presente sulla catena laterale o della lisina o dell'arginina; a differenza dell'acetilazione, questa non altera la carica netta del residuo interessato. Dei quattro istoni del core, sicuramente H3 risulta quello più pesantemente metilato, seguito da H4. I residui di arginina possono essere mono- o di-metilati, e in quest'ultimo caso possono esistere due configurazioni alternative caratterizzate da una modificazione simmetrica o asimmetrica. La lisina può accettare uno, due o tre gruppi metilici che portano alle corrispondenti forme mono-, di- o tri-metilate. Attualmente sono noti 24 siti di metilazione degli istoni (17K e 7R) che, considerando i tre possibili modi alternativi di modificazione di ciascun residuo, possono, in linea teorica, generare 3×1011 diverse isoforme di metilazione. Anche se è possibile che le cellule non utilizzino tutte queste forme, è particolarmente interessante comprendere il ruolo della metilazione e perché si siano evolute così tante diverse possibili varianti.
A differenza dell'acetilazione che, quasi senza alcuna eccezione, determina attivazione genica, la metilazione degli istoni può portare sia a induzione sia a repressione trascrizionale; questo dipende sia dal residuo modificato sia da altri cambiamenti simultaneamente presenti sugli istoni. La metilazione delle arginine, catalizzata da enzimi noti come 'arginina metiltransferasi' (PMRT, Protein arginine methyltransferase) sembra essere solamente presente sui geni attivamente trascritti; pertanto, questa modificazione è stata collegata all'attivazione trascrizionale. Al contrario la metilazione della lisina, dovuta principalmente all'attività di enzimi caratterizzati da un comune dominio catalitico di 140 amminoacidi, noto come SET, può avere diverse conseguenze funzionali, quali l'induzione dell'espressione genica, il suo silenziamento, la formazione dell'eterocromatina (quella regione della cromatina che rimane altamente condensata e trascrizionalmente silente per tutto il ciclo cellulare) e, infine, la perdita dei cromosomi. Inoltre, per quel che riguarda questa modificazione, inizia ad apparire chiaro come diversi distretti cromatinici siano caratterizzati da istoni diversamente metilati. Così, per esempio, l'eterocromatina pericentromerica è caratterizzata dalla presenza di un istone H3 tri-metilato sulla lisina in posizione 9 (K9) e da un H4 trimetilato su K20, mentre quella presente sul cromosoma X inattivo è arricchita dell'istone H3 tri-metilato su K27. Allo stesso modo, non tutta l'eucromatina (quella struttura poco impaccata e competente per la trascrizione) è uguale; per esempio, i geni potenzialmente attivi sono 'marcati' da istoni H3 e H4 rispettivamente mono-metilati su K36 e K4, mentre la loro effettiva attivazione sembra accompagnata da eventi di tri-metilazione.
Fino a poco tempo fa si pensava che la metilazione degli istoni fosse un processo irreversibile. Questo dogma derivava principalmente dall'osservazione che le lisine metilate nei nucleosomi hanno la stessa vita media degli istoni che li compongono, mentre i residui fosforilati e acetilati sono contraddistinti da un veloce turn over. Tale osservazione poteva essere spiegata assumendo che le modificazioni transienti avessero un ruolo regolativo, mentre la metilazione funzionava come un marchio permanente. A supporto di ciò vi era anche il fatto che, nonostante gli intensi sforzi, non era mai stato trovato un enzima capace di revertire questa modificazione post-traduzionale. Le nuove tecniche di biologia molecolare (ChIP, Chromatin immunoprecipitation) capaci di 'fotografare' in vivo la presenza sul DNA di una specifica proteina, o di una sua isoforma modificata post-traduzionalmente, hanno però rivelato come in alcune specifiche condizioni la metilazione degli istoni diminuisce velocemente nel tempo, rendendo quindi possibile l'esistenza di una demetilasi proteica. A conferma di ciò, negli ultimi anni sono state identificate le prime demetilasi ed è stato dimostrato come la rimozione del gruppo metilico dagli istoni possa avvenire attraverso diversi meccanismi. In particolare, la rimozione del metile dall'arginina sembra passare attraverso un processo che porta a un istone contenente una molecola di citrullina. Questo processo è catalizzato da una peptidil-arginina deiminasi (PADI4), incapace di agire sulle arginine demetilate.
In accordo con il legame tra metilazione delle arginine e attivazione trascrizionale, l'attività di PADI4 sembra essere riconducibile a meccanismi di silenziamento dell'espressione genica. Rimane ancora poco chiaro se la presenza nella cromatina di citrullina sia a sua volta un segnale capace di modulare la struttura della cromatina; inoltre, non è chiaro come la citrullina venga revertita ad arginina o metil-arginina. Per quel che concerne la demetilazione delle lisine, fino a ora è stato isolato un solo enzima, LSD1 (Lysine-specific demethylase 1), specificamente attivo sulla lisina 4 di H3; in accordo con il ruolo attivatorio di tale specifica modificazione, questa demetilasi era già stata individuata in precedenti complessi caratterizzati da funzioni repressive. LSD1 è incapace di demetilare residui tri-metilati; restano quindi ancora da individuare gli enzimi in grado di togliere questo marchio dalla cromatina.
La fosforilazione è un'altra importante modificazione covalente degli istoni, che sembra interessare prevalentemente la serina 10 di H3, portando a effetti completamente opposti. Infatti, la fosforilazione di S10 è necessaria sia per la condensazione dei cromosomi in mitosi sia per l'attivazione trascrizionale. Questa duplice e contraddittoria funzione della stessa modificazione può essere capita solamente imparando a non leggere ogni modificazione istonica come evento singolo isolabile dal contesto; infatti, l'effetto biologico di una modificazione dipende anche da quali altre modificazioni sono presenti sulla stessa proteina o sulle proteine appartenenti al medesimo nucleosoma. Così, tornando alla fosforilazione di S10, nell'induzione dell'espressione genica questa favorisce la successiva acetilazione di K14 della stessa coda istonica; al contrario, la condensazione mitotica avviene quando H3 è fosforilato non solo su S10 ma anche su S28 e T11. Per completezza, vogliamo ricordare che il gruppo amminico delle lisine degli istoni può anche essere modificato mediante l'attacco di peptidi noti come 'ubiquitina' o SUMO. Di queste modificazioni, così come delle ADP ribosilazioni degli istoni, per ora si conosce molto poco. In generale, riteniamo che l'ubiquitinazione degli istoni sia associata all'attivazione dell'espressione genica; al contrario, il legame di SUMO sembra essere implicato nella repressione trascrizionale. Da quanto detto, dovrebbe risultare chiaro come la presenza di una modificazione su un residuo istonico possa essere richiesta, possa favorire o bloccare quella successiva; pertanto, l'iniziale reclutamento di diverse HAT o HMT (istometiltransferasi) influenza la specifica combinazione di modificazioni che contraddistingue ogni locus genico (fig. 6A).
Ma come fa lo specifico pattern di modificazioni istoniche presente su un locus genico a esercitare la propria funzione regolativa? I vecchi modelli, derivati principalmente da studi sull'acetilazione, proponevano che le modificazioni post-traduzionali delle proteine del core avessero prevalentemente una funzione strutturale, capace di modificare o l'interazione del DNA con l'ottamero istonico, o la compattazione di una serie di nucleosomi. Si assumeva per esempio che tutte le modificazioni che diminuiscono la carica di un residuo, come l'acetilazione e la fosforilazione, riducessero il numero d'interazioni elettrostatiche tra il DNA e il core istonico, portando, di conseguenza, a una cromatina aperta e, quindi, trascrizionalmente accessibile. Oggi sappiamo che questa visione è troppo semplicistica e che, in vivo e in vitro, il DNA si avvolge attorno a ottameri formati da proteine iperacetilate con modalità del tutto simili a quelle riscontrate nei nucleosomi non modificati. Pertanto, la maggior parte degli addetti ai lavori ritiene che le modificazioni degli istoni influenzino la trascrizione, perché funzionano come punto di aggancio di specifiche proteine non istoniche capaci di modulare la struttura della cromatina e, quindi, di influenzarne le proprietà regolative.
In accordo con ciò, recentemente sono stati isolati specifici domini proteici che funzionano proprio come moduli di legame a istoni modificati (fig. 6B). Per esempio, i bromodomini sono deputati a legare selettivamente le lisine acetilate, mentre gli stessi residui metilati possono venire riconosciuti per lo meno da tre motivi proteici: i cromodomini, i domini Tudor e quelli contenenti ripetizioni WD40. In genere, ogni proteina si lega selettivamente solo a un particolare residuo istonico modificato. Per esempio, il cromodominio della proteina eterocromatica HP1 interagisce specificatamente con la lisina 9 di-metilata di H3, mentre quello della proteina polycomb (PC) si lega a K27 di-metilata. La situazione viene ulteriormente complicata e specificata dalla capacità delle modificazioni post-traduzionali, presenti sui residui vicini, di influenzare il legame di queste proteine. In sintesi, si evince come le code degli istoni rappresentino una piattaforma utile per stabilire diverse interazioni proteina-proteina e come il risultato finale di queste associazioni sia una particolare e ben controllata attività biologica del dominio cromatinico. In altre parole, si inizia a pensare che le diverse combinazioni delle modificazioni degli istoni stabiliscano un 'codice' (codice istonico) che, interpretato da altre proteine non istoniche, costituite da specifici domini d'interazione proteina-proteina, condurrebbe a uniche risposte cellulari.
Le varianti istoniche
Gli istoni del core sono codificati nella maggior parte degli organismi da diverse copie geniche, tutte altamente conservate e trascritte durante la fase di sintesi del DNA, quando il genoma duplicato deve essere opportunamente condensato. L'elevata conservazione di queste proteine suggerisce una loro capacità di legare il DNA in maniera aspecifica, indipendente dalla provenienza dell'acido nucleico. Questa aspecificità è molto importante per garantire la condensazione e il conseguente corretto funzionamento dell'intero genoma; l'insorgenza di mutazioni potenzialmente capaci d'influenzare le sequenze di DNA legate dagli istoni, è un evento estremamente pericoloso per la cellula, e quindi non selezionato favorevolmente nell'evoluzione. Recenti studi hanno dimostrato come una certa variabilità istonica sia stata ottenuta dai genomi eucariotici attraverso l'acquisizione di nuovi geni, codificanti per delle varianti istoniche, caratterizzate da piccoli cambiamenti nella sequenza amminoacidica e da una ristretta o ben definita distribuzione nella cromatina nucleare. In genere, queste varianti istoniche sono codificate da geni presenti in singola copia, la cui espressione non è limitata alla fase in cui viene replicato il DNA, ma estesa a tutto il ciclo cellulare. Alcuni di questi istoni si scambiano con quelli del core, costituenti la maggior parte della cromatina, durante lo sviluppo o il differenziamento portando a un'eterogeneità strutturale della fibra che può influenzare diverse funzioni nucleari quali la trascrizione, la segregazione cromosomica, la replicazione, la riparazione e la ricombinazione del DNA. Risulta quindi chiaro come l'evolversi di queste diverse forme istoniche abbia portato a un vantaggio aggiuntivo, la possibilità di assemblare dei nucleosomi con funzioni altamente specializzate e con una distribuzione nel genoma finemente controllata.
Per quel che concerne il numero e il tipo di varianti istoniche, sembra che l'evoluzione abbia favorito la divergenza delle proteine nucleosomiali più accessibili e, quindi, facilmente sostituibili. Infatti, mentre per H4 non è nota alcuna variante e H2B presenta solo poche varianti importanti durante la spermatogenesi, esistono quattro diverse forme sia di H2A sia di H3. Delle quattro forme di H3, H3.1, H3.2 e H3.3 appaiono molto simili, mentre CENPA, l'istone H3 centromerico, presenta una grande variabilità amminoacidica tra le diverse specie. Gli studi più recenti dimostrano come H3.3 sia caratterizzata da modificazioni post-traduzionali tipiche dei geni espressi; in accordo con ciò, questa variante viene riscontrata a livello delle sequenze attivamente trascritte. H3.3 differisce dalla forma H3 canonica solo per quattro residui, di cui tre presenti nella regione globulare. Questi ultimi permettono alla variante di associarsi con complessi proteici che la sostituiscono con il normale istone del core, mentre la RNA-polimerasi scorre sul templato per la sintesi del corrispondente trascritto. L'analisi della sequenza di CENPA ha dimostrato la presenza di un dominio globulare molto simile ad H3 e di una regione N-terminale completamente diversa. CENPA si localizza a livello del centromero ed è assolutamente essenziale per la sua formazione e il suo corretto funzionamento; in accordo con questa vitale funzione, l'eliminazione del gene codificante per CENPA in topi modello non permette lo sviluppo dell'animale. Benché non siano ancora noti i meccanismi molecolari responsabili della corretta localizzazione di questa proteina a livello del centromero, diversi esperimenti di biologia molecolare hanno dimostrato come, anche in questo caso, per un corretto reclutamento sia necessario il dominio globulare.
Per quel che concerne le varianti di H2A, sicuramente H2AZ rappresenta quella più studiata dal punto di vista funzionale. La sua importanza nei Mammiferi è testimoniata dal fatto che la distruzione dell'unico gene codificante per H2AZ non permette all'embrione in via di sviluppo di superare lo stadio della gastrulazione. Inaspettatamente, la stessa proteina sembra giocare un ruolo sia nell'attivazione sia nella repressione trascrizionale. Per quel che riguarda la proteina H2AX, invece, gli studi eseguiti su topi transgenici rivelano la sua importanza per la stabilità genomica e la fertilità maschile. In accordo con ciò, H2AX, distribuita in modo casuale sull'intero genoma, dove costituisce il 10÷15% dei nucleosomi, viene selettivamente fosforilata in seguito a rotture del DNA. Questo evento rappresenta il primo passaggio essenziale all'attivazione di un complicato meccanismo molecolare che, in ultima analisi, permette di riparare quei danni della doppia elica che sarebbero altrimenti letali per la cellula. Un'altra variante degna di menzione è la forma macroH2A. Questa, tipica solo dei Vertebrati, è caratterizzata da una coda N-terminale simile a quella presente su H2A e da un dominio C-terminale completamente diverso dagli istoni. MacroH2A è preferenzialmente localizzata sul cromosoma X inattivo. È interessante osservare che la presenza di macroH2A è un tipico marchio, ma non è essenziale all'inattivazione del cromosoma X. Riassumendo, benché ancora molti studi siano necessari per comprendere quante sono le varianti istoniche, come vengono depositate nella fibra cromatinica e qual è la loro funzione, possiamo affermare che esse si sono evolute per assolvere funzioni molto particolari; la loro inclusione nella struttura dei cromosomi porta a una maggiore dinamicità e alla formazione di domini cromatinici caratterizzati da nuove e specifiche proprietà.
I complessi di rimodellamento della cromatina nella regolazione dell'espressione genica
Diversi dati dimostrano che i complessi di rimodellamento della cromatina e quelli deputati a modificare post-traduzionalmente gli istoni cooperano funzionalmente. Per esempio, è noto come sia le mutazioni delle subunità del complesso SAGA (ad attività istonacetiltransferasica), sia quelle del complesso ATP-dipendente SWI/SNF di per sé non causano importanti difetti nella vitalità cellulare, ma, quando combinate insieme, conducono alla morte cellulare. Un simile risultato può essere spiegato solo ipotizzando un'interazione funzionale tra le due attività. Coerentemente con questi dati, è stato anche possibile dimostrare come sugli stessi promotori nelle cellule di mammifero possono essere contemporaneamente presenti entrambe le funzioni. In genere, si ritiene che la modificazione della cromatina a opera di un complesso possa facilitare l'attività dell'altro. In altre parole, su alcuni geni il rimodellamento ATP-dipendente potrebbe rendere più accessibile alle modificazioni post-traduzionali le proteine istoniche, mentre su altri loci i nucleosomi con code modificate influenzerebbero il legame di attività simili a SWI/SNF.
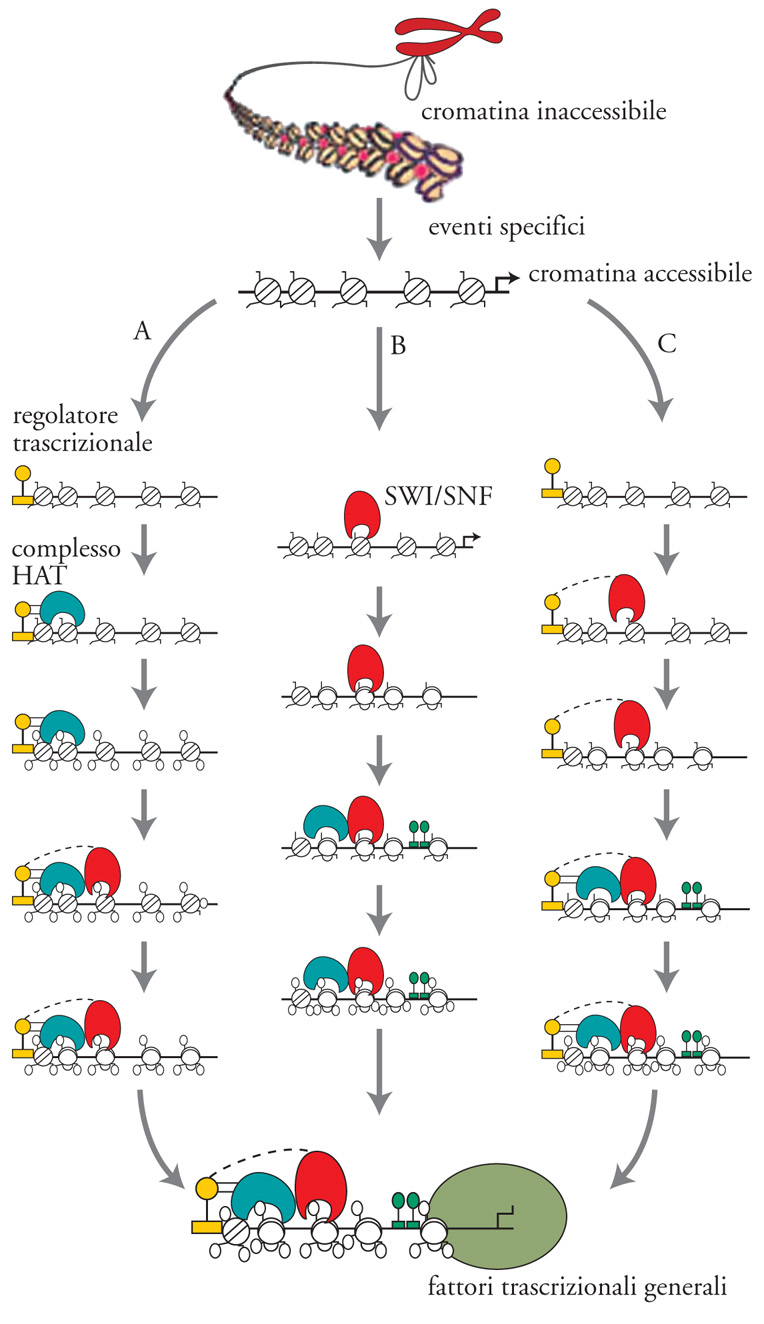
Quindi, anche se probabilmente la corretta regolazione trascrizionale di ciascun promotore necessita di entrambe le funzioni, non sembra esserci un ordine preferenziale nel loro utilizzo; ogni promotore lavora seguendo un proprio schema individuale avvalendosi di specifici complessi di rimodellamento, che possono differire per numero, tipo oppure ordine di intervento da quelli usati da un altro promotore. Anche l'apparato trascrizionale partecipa alla regolazione della specifica espressione di un gene, complicando ulteriormente la fitta rete di interazioni molecolari che si susseguono su un promotore. Così, per esempio, in alcuni casi il legame di un attivatore trascrizionale o il passaggio della RNA-polimerasi facilitano l'apertura della cromatina e il conseguente accesso delle attività di rimodellamento, mentre altre volte sono proprio le proteine che modulano direttamente la struttura e/o la distribuzione dei nucleosomi ad agevolare l'accesso del macchinario trascrizionale. In sintesi, la coordinata azione di tutte queste diverse funzioni permette al DNA stampo di raggiungere al momento giusto quell'architettura proteica, che gli garantisce il corretto livello di trascrizione (fig. 7).
La cromatina nelle patologie umane
Da quanto detto emerge come la fibra cromatinica non sia formata da un semplice susseguirsi di nucleosomi tutti uguali tra loro; al contrario, questa è composta da molteplici domini funzionalmente specializzati, distinguibili tra loro per la distribuzione e la densità nucleosomiale, per la presenza di specifiche modificazionipost-traduzionali o di varianti istoniche, e per l'associazione con proteine diverse da quelle presenti nei nucleosomi. Questa complessa serie di informazioni contenute nella cromatina permette di arricchire l'informazione insita nel codice genetico (DNA), alterando in modo stabile e potenzialmente ereditabile dalle cellule figlie l'espressione di un gene, senza modificarne la sequenza nucleotidica (epigenetica). Nel loro insieme i diversi meccanismi epigenetici, che comprendono principalmente la struttura della cromatina, con le sue modificazioni post-traduzionali e la presenza di diversi fattori proteici, e la metilazione del DNA permettono la formazione, negli organismi multicellulari, di tessuti e organi contenenti cellule diversamente differenziate nonostante il medesimo patrimonio genetico. L'importanza di questi meccanismi nel controllare la regolazione dell'espressione genica e, in ultima analisi, il destino di una cellula è dimostrato dal loro sempre più evidente coinvolgimento in diverse patologie umane. Infatti, molto spesso mutazioni a carico di complessi di rimodellamento della cromatina, capaci di modificarne il funzionamento o il reclutamento su specifici loci genici, portano a sindromi genetiche o allo sviluppo di tumori.
La natura sindromica di molti di questi disordini dello sviluppo può facilmente essere spiegata, se si ipotizza che la deregolazione della struttura cromatinica colpisce e altera l'espressione di più geni contemporaneamente. Un tipico esempio è dato dalla sindrome di ATR-X (Alpha-thalassemia/mental retardation) una rara patologia legata al cromosoma X, in cui i pazienti maschi affetti sono caratterizzati da un grave ritardo mentale, dismorfismi facciali caratteristici, microcefalia, anomalie ai genitali e una lieve α-talassemia. La causa molecolare di questa malattia risiede appunto in mutazioni a carico del gene ATRX, che codifica per una proteina facente parte di un complesso di rimodellamento della cromatina ATP-dipendente. Gli attuali modelli presuppongono che la patologia sia dovuta a una non preventivata trascrizione di geni normalmente repressi dal complesso multiproteico contenente ATRX. L'importanza delle attività di rimodellamento della cromatina nella biologia umana è ancora più evidente, se si considera il loro coinvolgimento nell'oncogenesi. Tra i meccanismi epigenetici che possono portare allo sviluppo di neoplasie, possiamo menzionare sia la repressione di geni, la cui attività è essenziale per contrastare lo sviluppo di un tumore, sia l'attivazione di geni normalmente silenti o meno espressi, la cui elevata trascrizione porta a un'eccessiva proliferazione cellulare. In generale, nel primo caso sono coinvolte attività quali le istondeacetilasi, le DNA- o le istonmetiltransferasi, e i complessi SWI/SNF, mentre nel secondo caso risultano coinvolte le HAT, le HMT e ancora SWI/SNF.
Bibliografia
Bannister, Kouzarides 2005: Bannister, Andrew J. - Kouzarides, Tony, Reversing histone methylation, "Nature", 436, 2005, pp. 1103-1106.
Fan 2003: Fan, Heng-Yu e altri, Distinct strategies to make nucleosomal DNA accessible, "Molecular cell", 11, 2003, pp. 1311-1322.
Guasconi, Ait-Si-Ali 2004: Guasconi, Valentina - Ait-Si-Ali, Slimane, Chromatin dynamics and cancer, "Cancer biology and therapy", 3, 2004, pp. 825-830.
Hansen 2002: Hansen, Jeffrey C., Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions, "Annual review of biophysics and biomolecular structure", 31, 2002, pp. 361-392.
Kamakaka, Biggins 2005: Kamakaka, Rohinton T. - Biggins, Sue, Histone variants: deviants?, "Genes and development", 19, 2005, pp. 295- 310.
Jenuwein, Allis 2001: Jenuwein, Thomas - Allis, C. David, Translating the histone code, "Science", 293, 2001, pp. 1074-1080.
Jiang 2004: Jiang, Yong-hui - Bressler, Jan - Beaudet, Arthur L., Epigenetics and human disease, "Annual review of genomics and human genetics", 5, 2004, pp. 479-510.
Lee 2005: Lee, David Y. e altri, Role of protein methylation in regulation of transcription, "Endocrine reviews", 26, 2005, pp. 147-170.
Martin, Zhang 2005: Martin, Cyrus - Zhang, Yi, The diverse functions of histone lysine methylation, "Nature reviews. Molecular cell biology", 6, 2005, pp. 838-848.
Narlikar 2002: Narlikar, Geeta J. - Fan, Heng-Yu - Kingston, Robert E., Cooperation between complexes that regulate chromatin structure and transcription, "Cell", 108, 2002, pp. 475-487.
Nowak, Corces 2004: Nowak, Scott J. - Corces, Victor G., Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation, "Trends in genetics", 20, 2004, pp. 214-220.
Peterson 2002: Peterson, Craig L., Chromatin remodeling enzymes: taming the machines, "EMBO reports", 31, 2002, pp. 320-322.
Peterson, Laniel 2004: Peterson, Craig L. - Laniel, Marc-André, Histones and histone modifications, "Current biology", 14, 2004, R546-551.
Sarma, Reinberg 2005: Sarma, Kavitha - Reinberg, Danny, Histones variants meet their match, "Nature reviews. Molecular cell biology", 6, 2005, pp. 139-149.
Woodcock, Dimitrov 2001: Woodcock, Christopher L. - Dimitrov, Stefan, Higher-order structure of chromatin and chromosomes, "Current opinion in genetics and development", 11, 2001, pp. 130-135.