
L'Ottocento: chimica. Atomi, tipi e radicali
L'Ottocento: chimica. Atomi, tipi e radicali
Atomi, tipi e radicali
Scoprire di che cosa sono fatti gli oggetti che ci circondano è la capacità fondamentale di un chimico. Tale tendenza umana universale ha in chimica un obiettivo raffinato, che rappresenta lo scopo stesso dell'analisi: questa, infatti, può avere una finalità immediata ‒ come determinare la composizione di un minerale, la purezza di un prodotto alimentare o l'identità di un farmaco ‒ oppure può riguardare questioni più sostanziali, quali la composizione, la forma tridimensionale e la struttura elettronica delle molecole o addirittura la determinazione degli isotopi degli elementi. A questo punto subentra il fisico, affrontando ulteriori questioni, come la struttura degli atomi e la natura delle particelle subatomiche.
Anche l'altra preoccupazione universale dei chimici, la sintesi, dipende dalla conoscenza della composizione: dagli alchimisti alle moderne società farmaceutiche la ricerca si è basata sempre su una più accurata comprensione della composizione chimica del prodotto desiderato. Malgrado quanto sostengano alcuni storici, fra i chimici vi è, e vi è sempre stata, una forte convinzione dell'oggettività delle ipotesi sulla struttura delle sostanze elaborate mediante gli apparati teorici e sperimentali della loro disciplina, e tale convinzione, avendo portato a risultati molto fruttuosi, si è rafforzata nel corso degli anni. In nessun altro campo, come nella chimica organica, la ricerca dell'oggettività è stata perseguita con altrettanta fiducia.
Dal punto di vista logico, il procedimento analitico inizia con l'identificazione dei costituenti di una miscela e prosegue con il ricavare le formule empiriche dei composti fino a quel momento non ancora conosciute, quindi le loro formule molecolari (in base alla conoscenza dei pesi molecolari), la disposizione nello spazio degli atomi costituenti (se vi è una qualche ambiguità) e poi la struttura fine in termini elettronici. Molto frequentemente, però, la sequenza storica delle scoperte non viene determinata dalla logica, ma piuttosto dal modo, il più delle volte accidentale, attraverso il quale si accumulano le conoscenze scientifiche.
Gli sviluppi storici possono essere riassunti in alcuni stadi, non sempre consequenziali, che saranno l'oggetto dei paragrafi seguenti.
Accettazione dell'atomismo chimico e dei pesi atomici
L'idea secondo la quale la materia potrebbe essere costituita da particelle è antica quasi quanto la nostra civiltà. A partire dai filosofi Greci, a più riprese si è sostenuto che l'operazione di dividere un oggetto in parti via via più piccole non potesse essere indefinitamente possibile; alla fine, infatti, si sarebbero ottenute particelle minuscole che non potevano essere ulteriormente suddivise. Queste ultime venivano denotate con vari nomi, come 'corpuscoli' o 'atomi'; di certo esse erano parte integrante della filosofia della materia di Newton e di altri filosofi meccanicisti del XVIII sec., sebbene nessuno, ovviamente, ne avesse mai ottenuta realmente una per poi poterla studiare. L'ipotesi delle particelle ultime della materia può successivamente essersi rivelata utile per speculazioni su fenomeni quali la diffusione gassosa, il moto browniano o anche per quelle sulla teoria cinetica dei gas, ma per un chimico la loro teorizzazione rivestiva un'importanza minore: in effetti, la nascita di un atomismo chimico doveva attendere l'opera di John Dalton (1766-1844) agli inizi del XIX secolo.
Dalton era un quacchero del North Country della Gran Bretagna, la cui teoria atomica fu formulata per la prima volta nel 1803 e poi presentata in modo più completo nel 1810. L'essenza della sua teoria consisteva nell'idea che differenti elementi chimici fossero costituiti da differenti tipi di atomi e che gli atomi dello stesso elemento fossero distinti da tutti gli altri da un peso atomico caratteristico. Tali masse erano troppo piccole per essere misurate direttamente e quindi si rese necessario introdurre una scala in cui il peso atomico di un elemento era calcolato in base al rapporto che sussiste tra il peso di uno dei suoi atomi e quello di un atomo di idrogeno (quest'ultimo assunto come unità di riferimento).
Il fatto che molti dei pesi atomici forniti originariamente da Dalton fossero sbagliati e la sua riluttanza a correggerli non possono oscurare la grandezza della sua intuizione. Il modo in cui egli vi arrivò non è ancora del tutto chiaro, sebbene siano state avanzate diverse ipotesi interpretative. Per un periodo di tempo egli visse nel Lake District, la parte più umida dell'Inghilterra, dove svolse studi sui fenomeni atmosferici che lo portarono a ragionare sul comportamento di miscele di gas, come l'aria, nella quale l'ossigeno e l'azoto non sembravano separarsi. Inizialmente pareva che in una miscela gassosa le particelle di un gas non respingessero quelle di qualsiasi altro gas, ma soltanto quelle dello stesso tipo, quindi ossigeno e azoto erano in grado di riempire completamente un recipiente che li contenesse entrambi. Di conseguenza, Dalton si chiese se gas diversi potessero avere particelle di dimensioni diverse, ma in tutte queste teorizzazioni gli atomi non venivano mai considerati come distanti l'uno dall'altro.
Gli studi sulla solubilità dei gas portarono Dalton a porsi il problema se le differenti solubilità potessero essere spiegate in termini di differenti dimensioni delle particelle. Sempre durante il primo decennio del XIX sec., Dalton aveva condotto esperimenti su diversi composti dello stesso elemento, quali i vari ossidi di azoto e gli idrocarburi come il metano e l'etilene. La sua conclusione ‒ cioè che tali composti erano diversi perché avevano proporzioni differenti, ma definite, dei loro costituenti ‒ era espressa in termini atomici, anche se gli storici continuano a discutere su cosa contò di più, se la comprensione teorica o la difficoltà nell'analisi dei dati sperimentali. Nel 1808 Dalton pubblicò il New system of chemical philosophy, completo di una tavola di 'nuovi' pesi atomici e di alcuni diagrammi di molecole semplici (perlopiù errati nel dettaglio, ma rivoluzionari per il loro impatto). Da quel momento, per esempio, sembrò possibile che i chimici potessero avere una conoscenza maggiore dell'ammoniaca, al di là del semplice fatto che essa contenesse 14 parti di azoto e 3 di idrogeno. Se quel rapporto di 14/3 (circa 4,7) era anche il rapporto tra i pesi atomici, allora una molecola di ammoniaca doveva contenere un solo atomo di ciascun elemento. In maniera analoga, si potevano scrivere 'strutture' per gli ossidi di azoto e per molti altri composti più comuni che avrebbero trasformato la percezione che i chimici avevano delle sostanze di laboratorio a loro più familiari. Agli inizi della elaborazione della teoria atomica ci si trova chiaramente di fronte ad argomentazioni che si potrebbero definire di tipo 'circolare': infatti, se l'ammoniaca era un semplice composto binario, allora certamente il peso atomico dell'azoto doveva essere 4,7. Di conseguenza, non si poteva usare questa deduzione per stabilire la formula dell'ammoniaca. Per sfuggire dal labirinto dell'argomentazione 'circolare' era necessario trovare dei metodi indipendenti per determinare i pesi atomici; questa fu la successiva tappa del viaggio verso la scoperta della struttura chimica delle sostanze.
Applicazione dei dati relativi ai pesi atomici per ottenere le formule empiriche
Uno degli aspetti più notevoli della storia della teoria atomica è che per oltre cinquant'anni molti chimici si rifiutarono di credervi: all'incirca per i primi vent'anni successivi al 1810, gli scettici erano la maggioranza. Le ragioni di questo rifiuto erano indubbiamente molte e complesse, ma due semplici fatti le sottendevano tutte. Il primo consisteva nella constatazione che nessuno fino ad allora aveva visto un atomo, e per i chimici inclini alla filosofia sensistica di John Locke, o addirittura a quella positivista in voga a Parigi, ciò bastava. Anche gli scettici, comunque, potevano divenire convenzionalisti e usare gli atomi come se essi esistessero, rimandando a un momento più opportuno il verdetto sulla loro effettiva esistenza. Il secondo fatto consisteva nella mancanza di accordo su quali fossero i pesi atomici delle sostanze elementari. Finché non si fosse saputo se il peso atomico dell'ossigeno era 8 oppure 16, diventava impossibile scrivere in modo sicuro persino una formula semplice come quella dell'acqua. Ciononostante ci si sforzava di sfuggire all'argomentazione 'circolare' e al pessimismo di quanti, nei loro calcoli sulla composizione chimica delle sostanze, ancora si limitavano ai pesi equivalenti (o di combinazione). Per prima cosa furono proposte alcune semplici regole empiriche. Dalton stesso ipotizzò che i più semplici 'atomi composti' binari (che oggi chiameremmo 'molecole') contenessero soltanto un atomo di ciascun elemento. Così, l'acqua avrebbe avuto la formula HO e il peso atomico dell'ossigeno sarebbe stato uguale a 8, dal momento che 8 parti in peso di ossigeno si combinano con 1 di idrogeno. Il chimico svedese Jöns Jacob Berzelius (1779-1848) andò oltre e suggerì che, nel caso in cui due elementi formassero più di una combinazione (come negli ossidi di carbonio o di azoto, o negli idrocarburi), allora essi si combinavano secondo i rapporti 1/1, 1/2, 1/3, e così via. Una volta trovato un accordo sul peso atomico dell'ossigeno, si poterono immediatamente determinare, a partire dalla composizione dei loro ossidi, quelli di molti altri elementi. Così, nell'ossido di carbonio (monossido di carbonio, CO) il rapporto in peso carbonio/ossigeno era 3/4; quindi, se O=8, ne conseguiva che C=6.
Una seconda via per la determinazione dei pesi atomici, indipendente dalla prima, sembrava trovare un fondamento nella legge scoperta nel 1809 da Joseph-Louis Gay-Lussac (1778-1850), riguardante i volumi di gas che si combinano chimicamente tra loro. La legge da lui enunciata stabilisce che i gas si combinano insieme secondo rapporti in volume espressi da numeri semplici e interi (assumendo che tutti i volumi siano, o possano essere, corretti alla medesima temperatura e pressione). Inoltre il volume del prodotto, se gassoso, sta anch'esso in rapporto semplice con quello dei costituenti, come è ben noto per quanto riguarda il caso dell'acqua: 2 volumi di idrogeno+1 volume di ossigeno=2 volumi di vapore acqueo.
Di lì a poco, nel 1811, una spiegazione rivoluzionaria fu avanzata dal fisico italiano Amedeo Avogadro (1776-1856). Egli sostenne che, nelle medesime condizioni di temperatura e pressione, volumi uguali di tutti i gas contenevano lo stesso numero di 'molecole'. Applicando quest'idea alla sintesi dell'acqua, potremmo concludere che 2 atomi di idrogeno (non uno) si sarebbero combinati con un atomo di ossigeno, il che avrebbe significato che la formula dell'acqua sarebbe stata (nell'attuale notazione) H2O, e il peso atomico dell'ossigeno sarebbe stato 16, non 8. Dalton, pur rimanendo affascinato da quest'idea, la rifiutò. Berzelius, invece, l'accolse entusiasticamente ed essa, quindi, aprì la strada alla determinazione di un'intera serie di pesi atomici corretti per i metalli e per altri elementi che si combinano con l'ossigeno. Negli anni successivi, infatti, la determinazione del maggior numero possibile di pesi atomici divenne uno dei principali obiettivi delle ricerche di Berzelius. In questo modo egli fornì un appoggio significativo allo sviluppo della teoria atomica, in un'epoca in cui molti altri chimici neanche la prendevano in considerazione.
In questa strategia fu supportato da altri due metodi, divenuti entrambi disponibili a partire dal 1819. Uno di essi derivava dalla legge dell'isomorfismo relativa alla forma dei cristalli enunciata dal chimico tedesco Eilhard Mitscherlich (1794-1863). Secondo tale legge due cristalli possono essere isomorfi, cioè avere la stessa forma geometrica, essendo ciascuno capace di crescere in una soluzione satura dell'altro. In tali casi, sosteneva Mitscherlich, l'analogia della loro forma esterna riflette una similarità della composizione interna: di qui la conclusione che questi cristalli avrebbero dovuto avere formule simili. Tra gli esempi addotti vi erano gli allumi e anche composti più semplici, come i vetrioli del ferro e dello zinco (le cui formule attuali sono FeSO4∙7H2O e ZnSO4∙7H2O). Se nel primo vetriolo era noto il peso atomico dello zinco, allora quello del ferro nel secondo sale si sarebbe potuto ricavare immediatamente.
Il secondo metodo, invece, derivava dalle contemporanee ricerche dei chimici francesi Pierre-Louis Dulong (1785-1838) e Alexis-Thérèse Petit (1791-1820), i quali, infatti, condussero un lungo studio sui calori specifici degli elementi solidi, concludendo che per molti di essi era valida la seguente relazione: peso atomico × calore specifico = ca. 6,4 (in termini attuali e in unità SI quest'espressione diventa: massa atomica relativa × calore specifico = 26). Dal momento che i calori specifici erano facilmente determinabili in laboratorio, l'utilizzazione della legge dei calori specifici, o legge di Dulong e Petit, rappresentava una maniera rapida e del tutto indipendente per calcolare i pesi atomici della maggioranza degli elementi allora noti. Anche in questo caso Berzelius fu in grado d'inserire i risultati di tali studi nel suo sistema generale di pesi atomici. Tuttavia, malgrado i suoi enormi sforzi, bisogna ricordare che per molti anni la teoria dei pesi atomici non ebbe successo. In ultima analisi, molte delle formule attribuite da Berzelius alle sostanze dipendevano ancora dal peso atomico dell'ossigeno, che si era convenuto dovesse essere 16 ed era a sua volta basato sull'assunzione dell'ipotesi di Avogadro. Tale accettazione tardò molto a realizzarsi, per motivi correlati all'estensione dell'ipotesi al problema della determinazione dei pesi molecolari.
Determinazione dei pesi molecolari e derivazione delle formule delle molecole
A partire dal 1825, in linea di principio fu possibile conoscere i pesi atomici della maggior parte degli elementi con sufficiente accuratezza (enormemente migliorata grazie alle tecniche di Berzelius) e ottenere quindi la formula empirica di molti composti più semplici: quella dell'acqua, per esempio, era H2O. Tuttavia, la situazione diventava confusa quando si passava a considerare i pesi molecolari. Dato il rapporto 2/1 tra idrogeno e ossigeno, una molecola d'acqua poteva essere espressa come H2O, H4O2, H6O3, H8O4, ecc., e non vi era modo di determinare quale di queste formule (se ve n'era una) fosse quella corretta. Questa limitazione costituiva un problema non tanto per i composti inorganici, quanto per quelli organici. Nel 1840 la situazione peggiorò, perché i lavori di Justus von Liebig (1803-1873) e di altri chimici avevano fornito un'analisi accurata di molti composti organici assegnandogli di conseguenza le formule empiriche corrette. I chimici compirono allora il successivo passo logico e determinarono non soltanto i rapporti degli atomi in un composto, ma anche il loro numero effettivo. È un'ironia della storia che, a quei tempi, le due sole strade valide per la determinazione dei pesi molecolari dei composti organici fossero entrambe bloccate dall'opposizione di quello stesso uomo che tanto aveva fatto per tenere viva la teoria atomica, cioè Berzelius.
Una prima obiezione riguardava l'uso che veniva fatto dell'ipotesi di Avogadro. Se essa veniva applicata anche ai prodotti della reazione oltre che ai reagenti iniziali, dalla sintesi del vapor d'acqua, per esempio, seguiva subito che 2 'atomi' di idrogeno + 1 'atomo' di ossigeno davano 2 'atomi' di vapor d'acqua. Di conseguenza, un solo 'atomo' di vapor d'acqua doveva provenire da 'mezzo atomo' di ossigeno ‒ il che era ritenuto impossibile. La difficoltà poteva essere aggirata supponendo che idrogeno e ossigeno fossero biatomici e usando per le loro particelle allo stato gassoso il nome di 'molecola'. In tal caso si sarebbe avuta la formulazione seguente, che coincide con quella oggi in uso: 2H2+O2=2H2O, dove il vapor d'acqua è H2O e non H4O2. È importante notare che in questo caso si fa ricorso a entità come H2 e O2. Analogamente, la reazione tra idrogeno e cloro, in cui un volume di ciascuno dei reagenti dà luogo a due volumi di acido cloridrico, può essere descritta e spiegata secondo la seguente equazione: H2+Cl2=2HCl.
Il concetto di 'molecola biatomica' di un elemento era però inaccettabile per Berzelius. Questi aveva associato la sua teoria degli atomi a un'assunzione basilare derivata dall'elettrochimica, e cioè che in una molecola gli atomi erano tenuti insieme da forze essenzialmente di tipo elettrico e ciascun atomo doveva mantenere invariata la propria polarità (positiva o negativa). Mentre la molecola HCl era ritenuta perfettamente possibile (con H positivo e Cl negativo), lo stesso non poteva essere sostenuto per H2, O2 o Cl2, in quanto molecole formate da due atomi uguali e quindi con la stessa polarità (evidentemente il legame omopolare non era ancora conosciuto). Come si è detto sopra, ciò scaturiva dal carattere sintetico della teoria dualistica di Berzelius, nella quale la parte corpuscolare era derivata da Dalton, mentre quella elettrochimica da Humphry Davy (1778-1829) e dalle sue ricerche sull'elettrolisi. Dunque, la sola posizione che Berzelius potesse adottare consisteva nell'accettare la proposta di Avogadro per gli elementi, ma non per i composti. Ciò sembrò illogico a molti chimici, ed è stato sostenuto che questa obiezione di carattere elettrochimico da parte di Berzelius sia stata uno dei maggiori ostacoli all'adozione generale dell'ipotesi di Avogadro, ma senza dubbio, oltre alla non irrilevante opposizione di Berzelius e di Dalton, esistevano altre ragioni. La situazione personale di Avogadro può aver giocato un ruolo, perché egli era tutt'altro che noto persino in Italia e le sue ricerche sulla teoria molecolare, pubblicate su una rivista francese, erano state ingarbugliate dalla loro estensione ai solidi oltre che ai gas e forse anche dalla sua incapacità di distinguere chiaramente tra 'atomi' e 'molecole'.
Un ulteriore discredito sulle ipotesi di Avogadro fu gettato quando il chimico francese Jean-Baptiste-André Dumas (1800-1884) misurò una serie di densità di vapore di differenti sostanze. Dal momento che queste rappresentano la densità di un gas rispetto a quella dell'idrogeno e assumendo che questo possieda la formula H2, era chiaro che si otteneva la relazione seguente: densità relativa (o del vapore) = 2 × peso molecolare. Quando però Dumas applicò questo tipo di prova ad alcuni elementi relativamente poco volatili, i suoi risultati sembravano suggerire formule come Hg2, I2, P4 e S6. Tali molecole poliatomiche erano ritenute del tutto improbabili, così che l'ipotesi di Avogadro subì un'ulteriore battuta d'arresto.
L'altro errore commesso da Berzelius riguardava il peso atomico dell'argento. Per determinare il peso molecolare di un acido organico egli lo convertiva nel suo sale d'argento; successivamente bruciava questo sale e misurava la perdita in peso. In questo modo era possibile calcolare quanti atomi di C, H e O erano associati a un atomo di argento e quindi ottenere la formula molecolare complessiva. Purtroppo egli assumeva che gli ossidi fortemente basici fossero biatomici, cosicché prendeva in considerazione formule (oggi ritenute errate) quali NaO, KO e AgO, che davano un peso atomico per l'argento pari al doppio di quello corretto. Di conseguenza, molte delle formule molecolari di composti organici indicavano il doppio rispetto al loro valore attuale. Alla fine l'errore sull'argento fu corretto applicando la legge di Dulong e Petit, e molti pesi molecolari sbagliati di composti organici furono ridivisi per 2 da Charles Frédéric Gerhardt durante gli anni Quaranta dell'Ottocento.
Malgrado tutto, è inevitabile pensare che dal 1810 al 1860 nella chimica regnasse un caos generalizzato, particolarmente nel campo della chimica organica. Quando Friedrich August Kekulé (1829-1896) pubblicò nel 1861 il famoso Lehrbuch der organischen Chemie (Trattato di chimica organica), egli incluse non meno di 19 differenti formule per una molecola di una sostanza assai familiare come l'acido acetico. Ciò dà una misura delle difficoltà in cui si trovarono i chimici fino alla fine degli anni Cinquanta del XIX secolo. Fortunatamente l'impasse stava per essere superata grazie al lavoro di Stanislao Cannizzaro (1826-1910), che risollevò le sorti dell'ipotesi formulata da Avogadro e mostrò in modo decisivo come l'applicazione coerente di tale ipotesi potesse dar luogo almeno a una rappresentazione veritiera della composizione molecolare delle sostanze organiche.
Il lavoro di Cannizzaro era intitolato Sunto di un corso di filosofia chimica e fu pubblicato nel 1858 sulla rivista "Nuovo Cimento"; nonostante fosse un resoconto del metodo di insegnamento di Cannizzaro presso l'Università di Genova, centrava il problema e offriva una chiara distinzione tra 'atomi' e 'molecole'. Ignorando le assunzioni semplificatorie di Dalton e di Berzelius, Cannizzaro mostrò come un'appropriata valutazione del lavoro di Avogadro, combinata con la vasta mole di risultati analitici accumulatisi a partire dal 1811, potesse portare direttamente a una strategia coerente finalizzata alla determinazione dei pesi atomici e molecolari delle sostanze. Questa era stata l'essenza del suo insegnamento presso un'università che inizialmente non aveva potuto neppure offrirgli un laboratorio attrezzato. Si potrebbe comunque sostenere che, in questo modo, egli non venne distratto nel suo lavoro teorico da incombenze amministrative e burocratiche che avrebbero potuto impedire lo sviluppo della sua ipotesi in un momento critico per la storia della chimica.
In effetti la crisi che si trovava ad affrontare la chimica in quel periodo era così grave che Kekulé e altri chimici pensarono di organizzare un congresso internazionale che si tenne nel 1860 a Karlsruhe. Cannizzaro partecipò alla conferenza e il suo intervento impressionò profondamente alcuni dei presenti, ma più importante fu il fatto che a ciascun partecipante fu data una copia del Sunto, e molti di loro, successivamente, sperimentarono un po' di quella sensazione di 'illuminazione' così classicamente espressa da Julius Lothar Meyer (1830-1895) al momento della lettura del testo di Cannizzaro.
Nel decennio successivo la chimica europea ebbe una rinascita; da quel momento in poi i pesi molecolari poterono essere accuratamente determinati e a ciascun composto si poté assegnare la propria formula molecolare caratteristica. Rimaneva ancora aperta, però, la questione della disposizione degli atomi nella molecola. La soluzione di questo problema fu fornita dalla comparsa, proprio in quel periodo, di un nuovo concetto, poi chiamato 'valenza'.
Prodromi del concetto di valenza
L'avvento di una nuova filosofia chimica e di mezzi affidabili per determinare i pesi atomici e molecolari non comportò l'immediata risoluzione del caos nel quale da lungo tempo si trovava la chimica organica. In realtà le cose dovevano ulteriormente complicarsi prima di iniziare a migliorare; occorre naturalmente del tempo perché le idee si diffondano attraverso l'intero sistema scientifico. Così, come si è detto, negli anni Sessanta dell'Ottocento circolava il manuale di chimica organica di Kekulé con non meno di diciannove formule diverse per l'acido acetico, e a Londra membri eminenti della Chemical Society dibattevano ancora seriamente circa l'esistenza degli atomi. Tuttavia, le cose si stavano muovendo e da due filosofie della chimica nettamente diverse emersero gradualmente idee che in ultima istanza si sarebbero fuse nel concetto successivamente definito con il termine 'valenza'. È impossibile ripercorrere ora nel dettaglio la storia di questo concetto, ci si limiterà solamente a menzionare gli aspetti più importanti: la 'teoria dei radicali' e la 'teoria dei tipi' che già contenevano in germe l'idea di valenza.
La teoria dei radicali
Come si è visto, Berzelius aveva elaborato una teoria elettrochimica della materia secondo la quale tutti i composti, organici e inorganici, erano tenuti insieme da forze di natura essenzialmente elettrica; in ogni molecola, dunque, vi erano una parte positiva e una negativa. Semplici composti inorganici quali l'acqua e l'acido cloridrico non ponevano alcun problema e, poiché un dato elemento era o positivo o negativo, secondo lo scienziato non potevano esistere entità come H2 oppure Cl2. Nei composti organici, però, uno o entrambi gli elementi potevano essere rimpiazzati da gruppi di atomi che rimanevano uniti, intatti, attraverso una serie specifica di reazioni chimiche; tali gruppi erano detti 'radicali'. In un primo momento, sull'esistenza reale di questi radicali vi fu un notevole e fondato scetticismo; nel 1832 Liebig e Friedrich Wöhler descrissero alcune esperienze nelle quali erano riusciti a isolare un 'radicale benzoile', sempre in combinazione con gruppi quali l'idrogeno, l'ossidrile, il gruppo amminico e quello ciandrico e altri gruppi negativi. Berzelius, è inutile dirlo, ne fu felicissimo.
Così come era possibile isolare il cloro dal suo composto con l'idrogeno, allo stesso modo doveva essere possibile isolare radicali quali il metile e l'etile dai relativi composti organici. Questo non fu possibile in un primo momento, sebbene dal 1837 al 1843 nel laboratorio di Robert Bunsen fossero state condotte ricerche su un radicale contenente arsenico, detto 'cacodile', che possedeva alcune proprietà peculiari: compariva in un gran numero di combinazioni con altri gruppi, ma lo si poteva trovare anche isolato; esso, quindi, fu il primo 'radicale libero' sulla scena della chimica. Ancora una volta Berzelius ne fu entusiasta e manifestò per lettera la propria approvazione a Bunsen. Oggi si sa che il radicale cacodile possiede la formula (CH3)2As, che il suo cloruro è (CH3)2 AsCl, mentre il suo ossido può essere scritto come [(CH3)2As]2O. In realtà quando Bunsen preparò il cacodile isolato egli non produsse il radicale (CH3)2As, ma il suo dimero [(CH3)2As]2. Comunque, a quel tempo, né lui né Berzelius erano assolutamente in grado di riconoscere la differenza fra il monomero e il dimero, e non vi è alcun dubbio che questo lavoro di Bunsen abbia esercitato una notevole spinta a favore della teoria dei radicali.
Tra i discepoli di Bunsen vi erano Adolf Wilhelm Hermann Kolbe (1818-1884) ed Edward Frankland (1825-1899). Il primo scoprì che l'elettrolisi dei sali di alcuni acidi alifatici dava luogo, all'anodo, alla formazione di un prodotto idrocarburico, e che l'acido acetico sembrava produrre il radicale metile. In realtà anche quest'ultimo era un dimero, l'etano; ma ciò non poteva essere rivelato da alcun esperimento allora accreditato e in ogni caso era contrario alle opinioni di Berzelius, che invece sembravano essere confermate da questi esperimenti. Lo stesso discorso valeva per il lavoro di Frankland sulle reazioni tra gli alogenuri alchilici e lo zinco. Lo ioduro di etile dava luogo a un prodotto idrocarburico e per alcuni mesi Frankland divenne noto e rispettato come lo scopritore del 'radicale etile'. Ora si sa con certezza che in realtà si trattava del butano, ma Frankland fu incoraggiato a produrre il maggior numero possibile di questi radicali, oltre che a esplorare un nuovo e vasto campo che gli si stava aprendo davanti, quello che egli denominò 'chimica metallorganica'. Dopo aver osservato molti nuovi composti dello stagno, dell'arsenico, dello zinco e di altri metalli, in una memoria intitolata On a new series of organic bodies containing metals pubblicata sulle "Philosophical Transactions", Frankland giunse a una conclusione importante:
Quando si considerano le formule dei composti chimici inorganici, anche un osservatore superficiale è colpito dalla simmetria generale della loro costruzione; i composti dell'azoto, del fosforo, dell'antimonio e dell'arsenico, soprattutto, mostrano la tendenza di tali elementi a formare composti contenenti 3 o 5 equivalenti degli altri elementi, ed è in queste proporzioni che le loro affinità sono soddisfatte nel modo migliore; così, nel gruppo ternario abbiamo NO3, NH3, NI3, NS3, PO3, PH3, PCl3, SbO3, SbCl3, AsO3, AsH3, AsCl3, ecc.; e nel gruppo a cinque atomi NO5, NH4O, NH4I, PO5, PH4I, e così via. Senza avanzare alcuna ipotesi sulla causa di questo raggruppamento simmetrico degli atomi, è sufficientemente evidente, dagli esempi ora forniti, che tale tendenza o legge prevale e che, a prescindere dalla natura degli atomi che gli si uniscono, la capacità di combinazione dell'elemento che li attrae, se mi si consente il termine, è sempre soddisfatta dallo stesso numero di questi atomi. (Frankland 1852, p. 440)
In questo lavoro, quindi, è contenuta la prima, chiara affermazione sulla "capacità di combinazione dell'elemento che attrae", cosicché si può sostenere che una parte importante della teoria della valenza era ormai definita. Tuttavia, non tutti ne erano convinti, e nel frattempo altri ricercatori affrontavano la questione da un'angolatura molto diversa.
La teoria dei tipi
La teoria dei radicali, come si è visto, era fondamentalmente dualistica, ma nel frattempo si andava sviluppando un punto di vista alternativo, la 'teoria dei tipi', definita dai suoi sostenitori anche 'ipotesi unitaria'. Tale teoria si interessava delle molecole nel loro complesso, senza separarle in ipotetici costituenti. I principali sostenitori di questo punto di vista erano i chimici francesi Auguste Laurent (1807-1853) e Gerhardt, e l'inglese Alexander W. Williamson (1824-1904), tutti attratti dal fascino della filosofia positivista di Auguste Comte che si diffondeva da Parigi e rifiutava qualsiasi forma di speculazione astratta o di superstizione. I sostenitori della teoria dei tipi aderivano a un empirismo molto spinto e si interessavano soltanto delle realtà verificabili sperimentalmente. Come in biologia, la cosa importante era confrontare, non analizzare. In un breve arco di tempo, gli anni Cinquanta dell'Ottocento, si assistette alla nascita di un certo numero di 'tipi' fondamentali a cui si potevano assegnare gli altri composti. Inizialmente i tipi principali erano tre: il 'tipo acqua', il 'tipo acido cloridrico' e il 'tipo ammoniaca'. Spesso essi venivano scritti nel seguente modo:
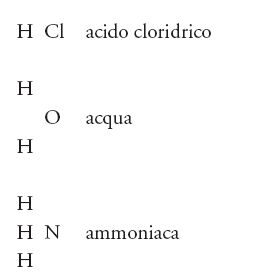
In base al loro comportamento chimico, molti composti potevano essere assegnati allo stesso tipo. Così, secondo Williamson, il tipo acqua includeva le seguenti sostanze:

mentre le nuove ammine scoperte da August Wilhelm von Hofmann (1818-1892) corrispondevano perfettamente al tipo ammoniaca. Compariva anche un 'tipo acido solforico' che con il pentacloruro di fosforo dava luogo alle seguenti reazioni:
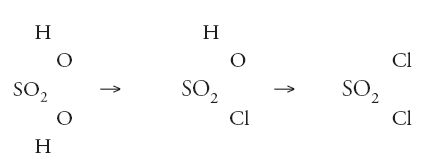
Da ciò non era difficile riconoscere che la formula dell'acido solforico poteva essere espressa come SO2(OH)2, o che in qualche modo il radicale SO2 univa a sé i due gruppi ossidrilici OH. In realtà, Williamson non arrivò ad affermare tanto, ma di lì a poco altri si mossero in direzione della elaborazione di un'ipotesi su quale tipo di forza di combinazione esistesse, emanante dal radicale SO2, e per mezzo della quale due, e soltanto due, gruppi come gli ossidrili potevano essere tenuti al loro posto nella molecola. Poco dopo a queste formule furono aggiunte delle parentesi graffe, che accrescevano l'idea di un qualche cosa di molto specifico che teneva unita la molecola:
Giunti a questo punto, non restava che un breve passo da compiere per arrivare al riconoscimento del tipo più importante per la chimica organica: il 'tipo carbonio'. Un chimico scozzese, Archibald S. Couper (1831-1892) vi andò vicino con una serie di articoli intitolati tutti On a new chemical theory e pubblicati alla fine degli anni Cinquanta dell'Ottocento. Egli scrisse le seguenti formule per gli alcoli metilico ed etilico:
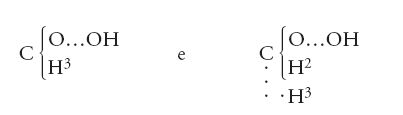
Sebbene Couper usasse gli attuali pesi atomici, tuttavia egli utilizzava l'eccentrico simbolo O…O, suo peculiare, per gli atomi di ossigeno; inoltre, il suo scopo era dimostrare come la teoria dei tipi fosse un'assunzione fallace dal punto di vista filosofico e priva di significato dal punto di vista chimico. Per questa ragione egli non era apprezzato dai teorici dei tipi, ma il suo articolo fu largamente ignorato per il semplice, ma tragico, motivo che poco dopo egli ebbe gravissimi problemi di salute e fu impossibilitato a pubblicare ulteriori lavori. Un'influenza assai maggiore fu esercitata invece dall'estensione della teoria dei tipi al tipo carbonio, per opera di Kekulé. Utilizzando per il carbonio il vecchio valore del peso atomico, nel 1857 egli introdusse un tipo 'gas delle paludi' (metano), scrivendo altri composti organici in modo simile, come, per esempio:

Nello stesso anno, Kekulé pubblicò un articolo, che diventerà molto famoso, riguardante la teoria dei radicali poliatomici, cioè quelli che, come l'SO2, sembravano tenere uniti diversi altri atomi. Egli suggerì che i radicali non fossero nulla più di ciò che rimaneva non legato in seguito a definite decomposizioni e concluse che la basicità di un radicale dipendeva dai suoi costituenti. Successivamente, nel 1858, scrisse un articolo intitolato Über die Constitution und die Metamorphosen der chemischen Verbindungen und die chemische Natur des Kohlenstoff (Sulla costituzione e le metamorfosi dei legami chimici e la natura chimica del carbonio), pubblicato negli "Annalen der Chemie und Pharmacie". Ritenendo necessario tornare agli elementi in quanto tali, egli giunse all'importante conclusione che nei radicali gli atomi di carbonio sono combinati tra loro secondo unità di affinità, mostrando che per n atomi di carbonio in un idrocarburo il numero di atomi di idrogeno deve essere pari a 2n+2, e che esistono serie omologhe in cui gli atomi di carbonio sono disposti nello stesso modo.
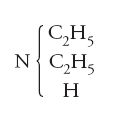
Kekulé era quasi arrivato alla meta e quattro anni più tardi rivendicò la sua priorità nell'introduzione del concetto di 'atomicità' (valenza) degli elementi. Ciò era vero in parte, perché già nel 1852, come si è visto, Frankland aveva chiaramente sostenuto almeno una parte di quella che in seguiro sarà definita 'teoria della valenza'. La cosa rilevante consiste nel fatto che sia Kekulé sia Frankland cercavano di unire gli aspetti più interessanti della teoria dei tipi e di quella dei radicali. Il secondo, in particolare, nel suo articolo del 1852, che segnò una discontinuità nell'evoluzione della teoria, arrivò addirittura a scrivere formule come:
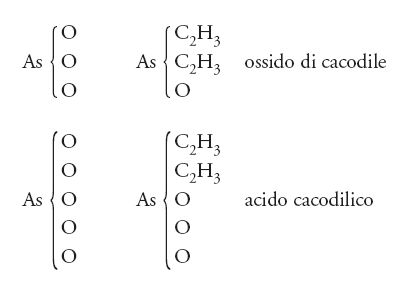
La valenza
Alla fine degli anni Sessanta dell'Ottocento i principali elementi di quella che sarebbe divenuta la teoria classica della valenza erano tutti definiti. Essi possono essere brevemente sintetizzati attraverso una serie di enunciati, risultato delle numerose ricerche sopra ricordate.
Il primo di questi enunciati sosteneva che ciascun elemento possedeva una definita capacità di combinazione. Questa, come abbiamo visto, era la conclusione di Frankland nel 1852. Per quanto ora essa possa apparire ovvia, ci volle mezzo secolo perché emergesse dal semplice atomismo di Dalton. Eppure, tale enunciato incontrò presto delle difficoltà, soprattutto perché Kekulé e alcuni suoi seguaci insistevano nell'affermare che la capacità di combinazione (comunque la si fosse chiamata) era, come il peso atomico, una caratteristica invariante di ogni elemento. Questa posizione aveva una certa logica, ma non spiegava molti fenomeni chimici ben noti, quali l'esistenza di due o più serie di cloruri dello stesso elemento (come ferro, rame e molti altri), o di reazioni quali: NH3+HCl=NH4Cl. In questo caso ci si poteva chiedere se la valenza dell'azoto fosse 3 o 5. Gli stratagemmi ideati dai sostenitori della valenza fissa per evitare tali domande erano numerosi; tra essi vi erano il concetto di 'composti molecolari' (secondo cui il cloruro di ammonio sarebbe stato in realtà NH3∙HCl), o quello di 'concatenamento' (secondo cui il cloruro del ferro bivalente sarebbe stato Cl2Fe‒FeCl2) o, infine, l'idea delle 'formule lineari' (secondo le quali l'acido solforico poteva essere rappresentato con l'improbabile formula H‒O‒O‒S‒O‒O‒H, dove lo zolfo sarebbe stato bivalente), e così via. Dopo molti anni fu concordemente accettato che, sebbene un elemento non potesse avere una valenza qualsiasi, spesso ne aveva più di una, e stava all'evidenza chimica, e più tardi fisica, determinare quale fosse quella appropriata.
Un secondo enunciato affermava che gli atomi polivalenti potevano accoppiarsi con altri costituenti di una molecola. L'accettazione di questa proprietà era dovuta principalmente a coloro che si collocavano nella tradizione della teoria dei tipi, in particolare Williamson e Kekulé. Questo assunto stava a significare che l'atomo polivalente in questione (N nell'ammoniaca, O nell'acqua, ecc.) poteva tenere uniti atomi come l'idrogeno e radicali come i gruppi alchilici. La teoria dei tipi, per citare un esempio, aveva indicato l'esistenza di somiglianze di famiglia tra le ammine, e ora si era trovato che la spiegazione risiedeva nella trivalenza dell'azoto. Una volta riconosciuto questo, la teoria dei tipi e le formule di cui essa si serviva potevano essere considerate ridondanti.
Un notevole progresso nella enucleazione dell'attuale teoria della valenza si ebbe, poi, con la scoperta della tetravalenza dell'atomo di carbonio. Sebbene Kekulé, Kolbe, Frankland e altri fossero impegnati in una feroce disputa su chi fosse il primo ad aver riconosciuto questa fondamentale verità della chimica organica, in realtà essi furono preceduti da William Odling (1829-1921), che nel 1855, in occasione di una conferenza presso la Royal Institution di Londra dal titolo On the constitution of hydrocarbons, introdusse un quarto tipo, CH4, da lui denominato 'tipo gas illuminante'. Tutti i composti radicalici dotati di esistenza autonoma (come quelli alchilici) potevano essere ridotti a un atomo di carbonio legato ad atomi di idrogeno o ad altri atomi di carbonio mediante le quattro unità di valenza che esso possedeva. Molti anni dopo, quando il fervore della controversia si era ormai spento, l'affermazione più onesta fu forse quella dello stesso Frankland: "L'applicazione della mia teoria della valenza agli atomi di carbonio, comunque, appartiene sostanzialmente a Kekulé" (1902, p. 189).
Una volta scoperta la tetravalenza del carbonio il passo successivo fu l'individuazione della possibilità che hanno gli atomi di alcuni elementi, specialmente il carbonio, di unirsi tra loro in catene. Due soli contendenti potevano rivendicare in maniera credibile di essere stati i primi a riconoscere il fenomeno del concatenamento: Couper e Kekulé. Tra loro, la palma spetta al secondo, non soltanto perché ne diede una enunciazione più chiara, ma anche per una questione di priorità temporale (nel 1858 in anticipo di poche settimane).
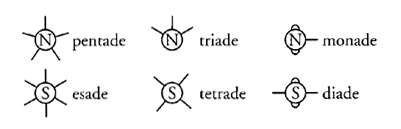
Un altro postulato fondamentale della teoria della struttura chimica dei composti organici affermava che in un composto chimico gli atomi erano legati tra loro mediante singoli legami di valenza. Il termine 'legame', nell'accezione di 'ciascuna unità capace di fissare gli atomi', fu introdotto da Frankland nella memoria Contributions to the notation of organic and inorganic bodies, pubblicata nel "Journal of the Chemical Society":
Con il termine legame, intendo fornire semplicemente un'espressione a ciò che è stato denominato dai differenti chimici in vari modi, come atomicità, capacità atomica ed equivalenza. Una monade è rappresentata come un elemento che ha un solo legame, una diade come un elemento che ha due legami, eccetera. È quasi superfluo sottolineare che con questo termine non intendo trasmettere l'idea di alcuna connessione materiale tra gli elementi di un composto: con ogni probabilità, i legami che realmente tengono uniti gli atomi di un composto chimico sono, per quanto riguarda la loro natura, molto più simili a quelli che tengono insieme i membri del Sistema solare. (Frankland 1866a, pp. 377-378)
Una versione di poco successiva di questo passo presente nelle Lecture notes for chemical students contiene una conclusione diversa: "I legami che realmente tengono uniti i costituenti, per quanto riguarda la loro natura, sono del tutto sconosciuti" (1866b [1870, I, p. 25]). Il semplice termine 'legame' valse più della maggior parte delle innovazioni a diffondere la nozione concreta di valenza ed è ancora in uso (per quanto non molti siano a conoscenza della sua origine). Esso si dimostra particolarmente efficace quando viene usato congiuntamente alle rappresentazioni grafiche dei legami stessi.
L'ultimo postulato, infine, sosteneva che la costituzione molecolare è rivelata nel modo più elegante dalla rappresentazione dei legami come linee. L'idea, oggi familiare, che i legami debbano essere rappresentati nelle formule mediante linee rette (come in H‒O‒H) risale a un fisico scozzese, Alexander C. Brown (1838-1922), che le utilizzò nella sua tesi di dottorato del 1861. Tre anni dopo, queste linee comparvero per la prima volta in un lavoro a stampa pubblicato nelle "Transactions of the Royal Society of Edinburgh". A partire dal 1866 di questa idea si era impadronito Frankland che la utilizzò nelle Lecture notes for chemical students. Le formule di Brown avevano una grande attrattiva euristica ed erano note come 'notazione di Frankland'. Tra gli esempi vi erano:
e i due cloruri di ferro, così rappresentati:
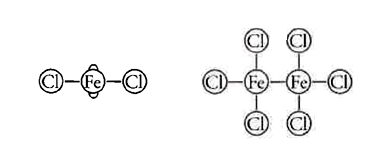
Poco dopo furono omessi i cerchietti. Sebbene Frankland continuasse a usare alcune formule di tipi per i composti organici, non ci volle molto perché esse sparissero del tutto e le moderne formule bidimensionali quasi-strutturali prendessero il loro posto. Il grande problema che sorse successivamente era se queste ultime rappresentassero davvero strutture reali e, in caso affermativo, in quale modo.
L'avvento della teoria della struttura
Per alcuni anni dopo il 1860 i termini 'formule di costituzione' o 'formule razionali' furono impiegati senza troppa precisione. Tuttavia molti problemi rimanevano da risolvere, come quello se tali formule rappresentassero lo stato permanente di una molecola o soltanto lo stato immediatamente precedente una reazione, oppure se fossero reali o solamente utili convenzioni o, infine, se la struttura (comunque venisse definita) fosse di importanza decisiva. Tra coloro che contribuirono a chiarire tali questioni vi furono Couper, Kekulé e Richard August Erlenmeyer, per citarne solo alcuni. Fu però il chimico russo Aleksandr Michajlovič Butlerov che nella memoria Die chemische Struktur der Stoffen (Sulla struttura chimica delle sostanze) espresse per primo la vera essenza della teoria della struttura: "Per ciascun composto è possibile una sola formula razionale, e quando saranno state derivate le leggi generali che governano la dipendenza delle proprietà chimiche dalla struttura chimica, questa formula esprimerà tutte queste proprietà" (Butlerov 1861, p. 559).
Negli anni Settanta dell'Ottocento i seguenti punti erano ormai accettati, sebbene non universalmente: (1) la formula di struttura di una molecola rivela semplicemente quali atomi sono legati a quali altri atomi, secondo le regole stabilite della valenza, e se sono coinvolti legami singoli, doppi o tripli; (2) la struttura di una molecola è una caratteristica peculiare e distintiva di quella molecola: gli isomeri, dunque, hanno differenti formule di struttura, sebbene condividano la stessa formula molecolare; (3) le proprietà di una molecola sono determinate in maniera cruciale dalla sua struttura; (4) la struttura è una caratteristica permanente di una molecola; (5) la struttura di una molecola può essere determinata mediante una serie di metodi fisici e chimici; (6) una struttura molecolare, così come i tipi che la precedettero, deve evidenziare alcune somiglianze 'di famiglia' tra composti di natura chimica simile.
La conversione delle semplici formule molecolari in formule di struttura fu un compito oneroso che la chimica organica dovette affrontare dagli anni Sessanta del secolo in poi. La scienza stava entrando nella fase in cui si potevano costruire alcune semplici regole fondamentali per la sintesi di un composto, potendo così fornire un'informazione definita sulla struttura del composto stesso. In modo analogo, l'analisi era in grado di fornire ulteriori chiarimenti, come nel caso dell'ozonolisi, in cui si rompe un doppio legame carbonio-carbonio e l'identificazione dei prodotti può mostrare il punto esatto in cui si trova il doppio legame in una lunga catena di atomi. La grande esplosione della sintesi organica verso la fine del XIX sec., soprattutto in Germania, portò a un accumulo di conoscenze sulla struttura di centinaia di prodotti naturali e sintetici.
Certamente, qualcuno nutriva ancora dei dubbi. Tra questi vi era Kolbe il quale, dopo aver ricevuto le Lecture notes di Frankland, liberamente arricchite con formule di struttura, temette che gli studenti potessero trarne troppe conclusioni (questo è un pericolo sempre insito in qualsiasi nuova modalità di rappresentazione scientifica); gli studenti, per esempio, potevano supporre che vi fosse una garanzia assoluta che gli atomi fossero disposti in quel modo, mentre in realtà esisteva sempre una componente probabilistica oltre, naturalmente, alla possibilità che alcune formule fossero semplicemente sbagliate. Tuttavia il pericolo più grande era che si potesse ritenere che tali rappresentazioni bidimensionali implicassero la bidimensionalità della molecola, come in effetti accadde, con risultati infelici. Questo particolare problema fu risolto con l'introduzione di quella che fu chiamata 'stereochimica'.
Gli inizi della stereochimica
La possibilità che le molecole potessero essere tridimensionali, anziché entità piatte, come implicavano le formule scritte sulla carta, era stata sempre presa in considerazione. Accenni a questo fatto si possono trovare nelle pagine di William H. Wollaston (1766-1828) e di Dalton, ma fino a quando non fu definita la teoria della struttura, tali concetti rimasero a un livello di mera speculazione.
Nel 1860 Louis Pasteur (1822-1895) era impegnato nello studio di ciò che poi divenne noto come 'attività ottica', vale a dire la capacità di alcune forme di un composto di ruotare il piano della luce polarizzata di un definito angolo in una certa direzione, e quella di altre forme di dar luogo allo stesso effetto, ma nella direzione opposta. L'acido tartarico aveva due di tali forme ('destro' e 'levo') e inoltre aveva una forma che non mostrava alcun effetto ottico. Pasteur si chiese se questo comportamento fosse correlato a una fondamentale asimmetria della molecola, tale che a livello molecolare una forma 'destro' potesse essere un'immagine speculare non sovrapponibile della forma 'levo' dello stesso composto. Se così era, l'isomeria ottica avrebbe rappresentato una violazione della legge fondamentale della teoria della struttura secondo la quale un composto poteva avere soltanto una data struttura molecolare. L'acido tartarico, per esempio, scritto HOOC∙CHOH∙CHOH∙COOH esisteva almeno in tre forme isomere. Queste idee furono sviluppate nel 1874 in un articolo di Joseph-Achille Le Bel, Relation entre la constitution chimique et le pouvoir rotatoire, nel quale si mostrava che in una molecola non planare esiste sempre una dissimmetria in composti del tipo Cabcd, ma se due qualsiasi di questi sostituenti sono tra loro identici (per es., se a=b), allora la dissimmetria sparisce. Stava ormai diventando disponibile un'abbondante quantità di evidenze sperimentali le quali dimostravano che le cose erano effettivamente così. Kekulé, nel frattempo, aveva ideato per scopi didattici un modello in cui un atomo di carbonio si trovava al centro di un tetraedro regolare e quindi i suoi legami di valenza erano non coplanari. L'ipotesi del carbonio tetraedrico fu proposta formalmente da Jacobus Henricus van't Hoff, nello stesso anno in cui fu pubblicato l'articolo di Le Bel, in un testo in fiammingo dal titolo Voorstel tot Uitbreiding der Tegenwoordige in de Scheikunde gebruikte Structuurformules in de Ruimte (Proposta per lo sviluppo di formule chimiche strutturali tridimensionali), che poi fu tradotto in francese in forma ampliata nel classico La chimie dans l'espace (1875). Tale ipotesi non spiegava solamente i fenomeni dell'isomeria ottica; essa poteva anche essere estesa a quella che venne presto denominata isomeria geometrica. Assumendo che in un doppio legame due atomi di carbonio sono rappresentati da due tetraedri aventi uno spigolo in comune, i quattro sostituenti sarebbero effettivamente coplanari con i due atomi di carbonio e potrebbero essere disposti in due modi diversi. Così, il caso apparentemente inspiegabile degli acidi maleico e fumarico poteva essere chiarito immediatamente:
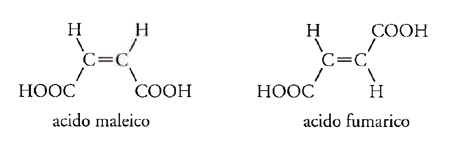
Questa descrizione comprendeva l'ipotesi che la rotazione attorno al doppio legame C==C fosse impossibile, mentre non sembrava esservi alcuna buona ragione per escludere la libera rotazione attorno a un legame singolo. Poco dopo queste idee furono estese al doppio legame C==N, chiarendo così molti altri casi di isomeria, come quella delle ossime della benzaldeide:
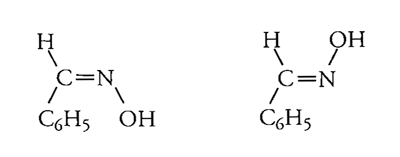
Molti di questi ragionamenti erano inaccettabili per i chimici della tradizione positivista e per altri, come Kolbe, che temevano un ritorno alla superstizione allora associata alla filosofia tedesca, nota come Naturphilosophie. Comunque, già nel 1880 essi appartenevano a una minoranza che diveniva sempre più esigua.
Infine bisogna notare che i legami di valenza direzionali (cioè diretti verso determinate direzioni) implicano un certo grado di tensione se sono distorti dal loro angolo naturale, che in un tetraedro regolare è di 109°128′. Da queste considerazioni Adolf von Baeyer (1835-1917) sviluppò la sua famosa 'teoria della tensione', mostrando che tale tensione sarebbe massima nel caso di atomi uniti da un doppio legame (circa 55°), ma esisterebbe anche in piccoli composti ciclici quali il ciclopropano e il ciclobutano, in cui gli angoli di legame sono rispettivamente 60° e 90°. Ciò aiutò a spiegare la reattività degli alcheni (nei quali il doppio legame determinava non una maggiore forza del legame, ma una sua maggiore debolezza) e la rarità di composti contenenti anelli di piccole dimensioni. Quando, verso la fine del XIX sec., alcuni di questi composti furono scoperti, la loro prevista instabilità fu confermata. Più rimarchevole, comunque, era l'esistenza di anelli tridimensionali. Sebbene ci si potesse aspettare che l'anello del cicloesano, che avrebbe dovuto avere un angolo interno di 120° nel caso fosse stato un esagono planare, mostrasse una certa tensione, nessuno aveva isolato due isomeri. Il lavoro di Hermann Sachse nel 1880 (largamente ignorato a quel tempo) e quello di Ernst Wilhelm Max Mohr nel 1919 misero in rilievo che la tensione poteva essere rimossa se l'anello fosse stato non planare, ma a forma di barca o di sedia:
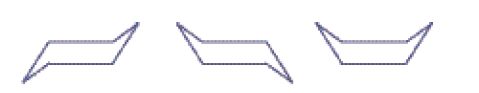
Queste entità strutturali erano note come 'conformazioni' e il loro studio veniva chiamato 'analisi conformazionale'. Tutto ciò non sostituiva la teoria della struttura ma la modificava, nella misura in cui si accettava: (1) una struttura tridimensionale per molte molecole; (2) il concetto, di validità generale, di 'rotazione libera' attorno a un legame singolo C‒C; (3) il riconoscimento che questa rotazione non poteva essere completamente libera nei casi in cui erano coinvolti sostituenti di grandi dimensioni, o laddove (come nel cicloesano) poteva essere ostacolata dalla formazione di un intermedio, tra un conformero e l'altro, che presentasse una certa tensione, o laddove un anello non planare fosse fuso con un altro, come nel decaidronaftalene. L'isolamento di due forme di quest'ultimo rappresentò la conferma definitiva dell'ipotesi conformazionale.
Bibliografia
Benfey 1966: Kekulé centennial, a symposium co-sponsored by the Division of History of Chemistry, the Division of Organic Chemistry and the Division of Chemical Education at the 150th meeting of the American Chemical Society, Atlantic City, N.J., Sept. 15-16, 1965, edited by Otto T. Benfley Washington, American Chemical Society, 1966.
Bradley 1990: Bradley, J., Cannizzaros Methode: der Schlüssel zur modernen Chemie, Bad Salzdetfurth, Franzbecker, 1990.
Brock 1967: The atomic debates. Brodie and the rejection of the atomic theory. Three studies, edited by William H. Brock, Leicester, Leicester University Press, 1967.
‒ 1992: Brock, William H., The Fontana history of chemistry, London, Fontana, 1992.
Brooke 1995: Brooke, John Hedley, Thinking about matter. Studies in the history of chemical philosophy, Aldershot-Brookfield, Variorum, 1995.
Cardwell 1968: John Dalton and the progress of science, edited by Donald S.L. Cardwell, Manchester, Manchester University Press; New York, Barnes and Noble, 1968.
Knight 1992: Knight, David M., Ideas in chemistry. A history of the science, London, Athlone, 1992.
Lundgren 1979: Lundgren, Anders, Berzelius och den kemiska atomteorin, Uppsala-Stockholm, Almqvist & Wiksell International, 1979.
Melhado 1981: Melhado, Evan M., Jacob Berzelius. The emergence of his chemical system, Stockholm, Almqvist & Wiksell International; Madison, University of Wisconsin Press, 1981.
Mierzecki 1991: Mierzecki, Roman, The historical development of chemical concepts, translated by Andrzej Diniejko, Warszawa, Pañst. Wydaw. Naukowe; Dordrecht-Boston, Kluwer Academic, 1991 (ed. orig.: Historyczny rozwój pojêae chemicznych, Warszawa, Pañst. Wydaw. Naukowe, 1985).
Morselli 1984: Morselli, Mario, Amedeo Avogrado. A scientific biography, Dordrecht, Reidel, 1984.
Palmer 1965: Palmer, William G., A history of the concept of valency to 1930, Cambridge, Cambridge University Press, 1965.
Ramsay 1975: Van't Hoff - Le Bel centennial, edited by O. Bertrand Ramsay, Washington, American Chemical Society, 1975.
Russel 1971: Russell, Colin A., The history of valency, Leicester, Leicester University Press, 1971.
Smyth 1997: John Dalton, 1766-1844. A bibliography of works by and about him, with an annotated list of his surviving apparatus and personal effects, 2. ed., edited by Albert L. Smyth, Manchester-Aldershot, Manchester Library and Phil. Pubns., in association with Ashgate, 1997 (1. ed.: Manchester, Manchester University Press, 1966; 3. ed., revised and expanded edition, Aldershot-Brookfield, Ashgate, in association with Manchester Library and Phil. Pubns. [1998]).
Stranges 1982: Stranges, Anthony N., Electrons and valence. Development of the theory 1900-1925, College Station, Texas A&M University Press, 1982.