
Immunopatologia
Immunopatologia
(App. V, ii, p. 622)
Gli aspetti più rilevanti nell'ambito dell'immunologia hanno riguardato, negli anni Novanta, varie malattie, con una migliore definizione sia della patogenesi sia dei meccanismi di controllo che regolano le alterazioni della risposta immunitaria in modelli sperimentali e in clinica. Aree di ricerca che hanno ricevuto particolare impulso sono: immunodeficienze congenite; reazioni di ipersensibilità (malattie allergiche); malattie autoimmuni. I progressi in questi settori hanno avuto implicazioni sia di ordine teorico, con l'approfondimento delle conoscenze di base nella fisiopatologia del sistema immunitario, sia nella terapia medica.
Immunodeficienze congenite
L'importanza dei deficit immunitari congeniti riguarda la comprensione dei fenomeni che permettono di interpretare le alterazioni selettive responsabili dell'anomalia considerata alla base del difetto diagnosticato. La prevalenza dei disordini da immunodeficienza congenita o primitiva (PID, Primary Immunodeficiency Diseases) varia in modo notevole in rapporto al tipo di malattia. Allo scopo di studiare meglio le PID è nato un registro europeo che raccoglie i dati su pazienti appartenenti a varie nazioni (al novembre 1998 sono state raccolte informazioni provenienti da oltre 24 paesi, tra cui l'Italia) e che rappresenta una preziosa banca dati sia per finalità epidemiologiche, sia per la corretta impostazione di analisi genetiche e per una razionale definizione nell'approccio terapeutico. La stessa Organizzazione mondiale della sanità (OMS) si giova di un comitato internazionale di esperti (Immunodeficiency Study Group) per un costante aggiornamento delle malattie da deficienza immunitaria. Le PID includono, secondo la classificazione OMS (1999), i seguenti raggruppamenti: deficit combinati; deficit prevalentemente anticorpali; forme ben definite di patologia associata a deficit immunologico; deficit delle frazioni complementari; deficit della fagocitosi (quantitativa e/o funzionale).
Per alcune di queste malattie l'interpretazione del danno immunitario si è chiarita grazie all'adozione di tecniche di genetica, di biologia molecolare e analisi di citologia funzionale.
SCID (Severe Combined Immunodeficiency). Appartiene al gruppo dei deficit combinati. Le SCID sono il prototipo delle PID a più drammatica estrinsecazione clinica. In tale situazione patologica l'organismo non è in grado di attivare né una risposta anticorpale né una risposta di tipo cellulo-mediato. Si possono distinguere due forme: una con difetto dei B e T linfociti, e l'altra nella quale i linfociti B possono essere normali o addirittura aumentati, ma con assenza dei T. Una più estesa comprensione dei difetti presenti nella cellula linfocitaria si è avuta con gli studi sull'adesione cellulare e sulla genesi dei segnali di transduzione necessari alla funzione della cellula stessa.
Infatti, quando un antigene viene esposto sulla cellula professionista APC (Antigen Presenting Cell) per la presentazione al linfocita reattivo, scattano su questa cellula una serie di segnali transmembrana con attivazione di molecole intracitoplasmatiche. Si osservano fenomeni di fosforilazione con il reclutamento di proteine chiave in grado di interagire a diversi livelli e dar luogo, in ultima analisi, alla produzione di citochine (per es. l'interleuchina-2, IL-2).
Nell'ambito della SCID legata al sesso (X-linked), nella quale mancano i T ma sono presenti i B, il meccanismo patogenetico riconosciuto riguarda alcune mutazioni sulla catena γc dei recettori per le interleuchine IL-2, IL-4, IL-7, IL-9 e IL-15. Nella forma autosomica recessiva (sempre a fenotipo T-B+) le mutazioni riguardano una chinasi intracellulare (JAK3), che si lega alla catena γc. Nell'ambito delle SCID causate da un particolare difetto enzimatico è stato proposto, all'inizio degli anni Novanta, l'impiego della terapia genica.
Agammaglobulinemia X recessiva
Originariamente segnalata come malattia di Bruton, questa forma morbosa risulta clinicamente caratterizzata da un'aumentata suscettibilità alle infezioni batteriche. È il prototipo delle PID a prevalente difetto anticorpale. Il sistema immunitario deputato alla risposta specifica umorale (linfociti B) nei confronti dell'antigene presenta una marcata incapacità di produrre anticorpi sia in seguito a infezioni sia dopo vaccinazioni. La diagnosi viene effettuata mediante determinazione delle immunoglobuline sieriche, che sono assenti o gravemente deficitarie in tutte le tre classi IgG, IgA e IgM. L'incidenza della forma morbosa ha una stima di circa 1:150.000 nati vivi. Sono peraltro noti anche casi che colpiscono il sesso femminile, con un quadro praticamente non distinguibile da quanto osservato nel bambino.
Il gene responsabile della XLA (X-Linked Agammaglobulinemia) risulta localizzato sul braccio lungo del cromosoma X (Xq22). Questo gene codifica per un enzima essenziale della differenziazione del linfocita B: tirosin chinasi (btk, dalle iniziali di Bruton tirosin chinasi). La molecola, formata da 659 aminoacidi, esplica una funzione importante nel transdurre i segnali che sono necessari alle fasi di proliferazione e differenziazione dei B linfociti. Esistono segnalazioni di forme 'atipiche' nelle quali la produzione di anticorpi e il numero dei B linfociti sono più elevati rispetto alla malattia classica. Sembra comunque che per entrambe le varianti l'alterazione dipenda dal gene che codifica per btk.
Per esempio, nel sesso femminile le cellule B hanno inattivazione del solo cromosoma X, che è andato incontro a mutazione. Le conseguenze di ricerche miranti a definire gli aspetti cellulari e molecolari dell'agammaglobulinemia legata al sesso hanno avuto immediata applicazione pratica in ambito diagnostico. Infatti, sebbene le indagini morfologiche sul linfonodo e funzionali sulle cellule (linfociti) del sangue periferico consentano di ottenere utili elementi per la diagnosi differenziale nei confronti di altre malattie congenite con carenza di anticorpi (immunodeficienza comune variabile, immunodeficienza con produzione aumentata di IgM), gli aspetti biologico-molecolari forniscono un definitivo inquadramento della forma morbosa.
Per una comprensione più completa della trasmissione della malattia all'interno di gruppi familiari che hanno uno o più membri colpiti, è utile eseguire l'analisi di linkage o di segregazione degli RFLP (Restriction Fragment Length Polimorfism, polimorfismo di lunghezza dei frammenti di restrizione). Con l'applicazione di queste metodiche di laboratorio è possibile identificare il cromosoma responsabile della patologia cercata. La clonazione del gene permette inoltre di valutare i caratteri propri della mutazione e ne consente un'analisi discreta, sostituendo in pratica la stessa applicazione del linkage.
L'opportunità offerta da una tempestiva diagnosi è importante non tanto perché la diagnosi differenziale con altre forme di ipogammaglobulinemia possa modificare un approccio terapeutico tempestivo (basato sulla terapia sostitutiva con immunoglobuline), ma perché rende possibile definire l'insieme dei rischi di trasmissione del difetto. Infatti, in una donna portatrice dell'anomalia il rischio di trasmissione al figlio è del 50%, ma se la mutazione riguarda la sola linea germinale, con comparsa del tipo sporadico, ne risulta un rischio scarsamente significativo per la trasmissione del difetto alla prole. Naturalmente l'indagine va estesa anche alle altre componenti femminili del gruppo familiare, in particolar modo per quanto riguarda le sorelle della madre con figlio affetto da agammaglobulinemia X-linked. Sebbene non si abbiano ancora elementi validi per attuare una terapia genica per questo difetto immunitario, la conoscenza del gene responsabile della XLA e delle alterazioni conseguenti ha offerto concrete possibilità di futuri progressi in questo settore.
Immunodeficienza con iper IgM
Si tratta di un raro difetto della sintesi anticorpale caratterizzato da ridotti livelli degli isotipi IgG e IgA, con valori delle IgM aumentati o presenti a concentrazione normale. Anch'esso rientra nei difetti a prevalente estrinsecazione anticorpale. Nell'ontogenesi del sistema immunitario la maturazione della linea cellulare B avviene (nell'adulto) all'interno del midollo osseo. Dalla cellula staminale pluripotente ha origine una sequenza di cellule che acquisiscono, attraverso passaggi successivi, le immunoglobuline di membrana IgM e IgD. Il linfocita B maturo, in grado cioè di rispondere allo stimolo antigenico, è IgM+IgD+. La produzione di anticorpi durante la risposta primaria è di tipo IgM. Quando i linfociti B memoria rispondono a successivi stimoli antigenici, gli isotipi di immunoglobuline cambiano e vengono sintetizzati anticorpi IgG o IgA (switch isotipico). Gli studi sull'interazione cellulare e sui meccanismi che regolano l'attivazione dei linfociti B hanno evidenziato l'importanza dei segnali che i linfociti T forniscono ai B per modulare l'attivazione della risposta immunitaria.
In particolare, nell'immunodeficienza X-recessiva con iper IgM è stato identificato un difetto causato da mutazione che riguarda il gene per la molecola CD40L espressa sui linfociti T CD4+ attivati. Il CD40L è una glicoproteina che possiede un breve tratto intracitoplasmatico, una regione transmembrana, e una componente all'esterno della cellula. Sui linfociti B si trova una molecola, definita controrecettore, identificata come CD40. Il linfocita T, combinando il proprio CD40L con il CD40, induce un'interazione che dà origine a segnali necessari per l'attivazione e la differenziazione dei linfociti B. D'altro canto il CD40 non è espresso solo dai linfociti B, ma anche da altri tipi cellulari tra i quali le cellule presentanti l'antigene (APC). In conseguenza di un'alterazione del CD40L, i linfociti B non possono evolvere verso gli stadi tardivi di maturazione e non si producono le cellule memoria, né si verifica lo switch isotipico. Poiché le cellule APC non interagiscono, per lo stesso motivo, con i linfociti T attivati, vengono meno i segnali di attivazione mediati dall'interleuchina 12 (IL-12) e non si osserva la produzione di IFN γ (interferone), che è uno dei primi importanti fattori di stimolo per le cellule fagocitarie e la loro funzione microbicida.
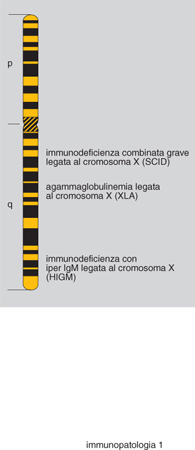
Clinicamente il quadro si caratterizza per infezioni a carico delle alte e basse vie respiratorie (di particolare gravità è la polmonite da Pneumocystis carinii), con fenomeni gravi di diarrea ad andamento cronico e conseguente ritardo della crescita. Varie mutazioni sono state identificate sul gene del cromosoma X (fig. 1), localizzato a livello Xq 26.3-27. Gli aspetti terapeutici includono l'impiego di immunoglobuline per via endovenosa e il trapianto di midollo osseo da familiare HLA-identico (HLA, Human Leukocyte Antigen).
Atassia-teleangectasia
Questa forma patologica rappresenta una caratteristica sindrome autosomica recessiva (patologia ben definita associata a deficit immunitario), caratterizzata da fenomeni progressivi di involuzione cerebellare, comparsa di fini teleangectasie, soprattutto evidenti alle sclere e, in circa due terzi dei casi, con immunodeficienza variamente espressa (difetto delle sottoclassi IgG2, IgG4, delle immunoglobuline IgA e delle IgE).
In particolare, è ridotta la risposta ad antigeni con struttura polisaccaridica, il numero dei linfociti T è alterato e si osserva un incremento della sintesi di autoanticorpi.
Lo squilibrio biologico riguarda vari aspetti del ciclo cellulare che si estrinseca, tra l'altro, in una maggiore suscettibilità al danno causato dalle radiazioni ionizzanti. Sono state dimostrate frequenti rotture cromosomiche, alterazioni dei geni per il TCR (T Cell Receptor) e anomalie sui cromosomi 7 e 14. In genere la morte dei pazienti con atassia-teleangectasia è causata da malattie infettive respiratorie o dalla comparsa di neoplasie maligne a carico del sistema linforeticolare. Il gene per questa sindrome è stato isolato nel 1995 e viene definito ATM (Atassia-Teleangectasia Mutation, per mutazione propria dell'A-T). Sono state descritte oltre 150 mutazioni. Il prodotto interessato è una molecola con forte omologia per la fosfoinositide-3-chinasi.
Per quanto concerne le immunodeficienze acquisite, v. immunodeficienza acquisita, sindrome da (AIDS), App. V, e il relativo aggiornamento in questa Appendice.
Reazioni di ipersensibilità
Nel corso di alcune reazioni immunitarie causate da particolari molecole (allergeni) si osserva l'attivazione di meccanismi effettori assai più forti e rapidi di quanto si osserva nel contesto di una risposta immunitaria normale. Una particolare attenzione ha ricevuto l'analisi delle reazioni di ipersensibilità mediata da anticorpi IgE. Infatti, sebbene sia da tempo nota la sequenza degli eventi che causano la produzione di questo isotipo di immunoglobuline (sensibilizzazione dei cloni linfocitari B; binding tra cellule con recettore per le IgE e allergene; interazione tra allergene e IgE adese sulle cellule effettrici; liberazione dei mediatori chimici presenti nelle cellule attivate), è rimasto per molto tempo non chiarito il motivo per cui alcuni comuni allergeni possono causare risposte del tutto sovrapponibili a quelle osservate verso determinati agenti patogeni, mentre altre molecole, introdotte con le stesse modalità degli allergeni, non inducono una sensibilizzazione mediata dagli anticorpi IgE. Studi di biochimica e biologia cellulare hanno contribuito a una migliore definizione del panorama.
In realtà, il problema è complesso perché, mentre risulta possibile che lo stato di allergene sia una caratteristica della molecola, tuttavia è altrettanto ragionevole supporre che esistano elementi 'aggiuntivi' in grado di guidare in qualche modo l'allergene verso la sintesi di IgE. In sostanza, se le IgE possono funzionare egregiamente nei confronti di un'infestazione parassitaria, facilitando l'espulsione dei parassiti che hanno invaso l'organismo, non è di immediata comprensione il meccanismo per cui la risposta verso alcune molecole di per sé non patogene (per es. i pollini) possa scatenare la liberazione di sostanze chimiche vasoattive in grado di danneggiare l'ospite, senza che ve ne sia una vera e propria necessità biologica.
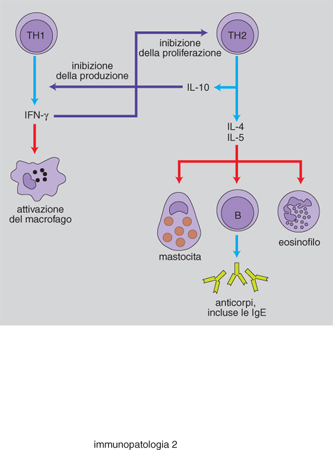
Molti studi hanno perciò riguardato i sistemi di regolazione della risposta IgE e le interazioni tra le cellule immunocompetenti. Sono stati in tal modo messi in evidenza aspetti di predisposizione genetica, le caratteristiche dell'antigene e le modalità di somministrazione; tuttavia un sostanziale passo avanti è stato compiuto grazie all'identificazione di cellule T helper che possono essere distinte in due sottopopolazioni (TH1 e TH2). Varie osservazioni hanno consentito di dimostrare come le cellule TH2 siano preferenzialmente coinvolte in alcuni individui, durante la risposta a particolari antigeni. Le TH2 sintetizzano citochine che facilitano una risposta di tipo allergico: IL-3 e IL-4 stimolano la proliferazione dei mastociti e la IL-4, in particolare, svolge un ruolo essenziale nell'indurre la produzione di IgE da parte dei linfociti B attivati. La conseguenza consiste nel fatto che vengono prodotte molte IgE, piuttosto che immunoglobuline di altro isotipo come le IgG o le IgA. D'altro canto un'altra interleuchina prodotta dai TH2, la IL-5, di per sé agisce favorendo la maturazione e la produzione degli eosinofili, che svolgono un ruolo importante nella genesi delle reazioni allergiche. L'equilibrio quindi risulta spostato verso un trend funzionale che privilegia un sistema 'allergizzante' rispetto, per esempio, a quanto si verifica con l'attivazione delle TH1 che producono citochine (quali l'IFN-γ e IL-2) con azione inibitoria sulla risposta di tipo IgE (fig. 2).
Sul piano clinico la produzione di IgE determina i fenomeni di ipersensibilità immediata poiché la sequenza degli eventi è molto rapida e i mediatori chimici che intervengono sui vari substrati anatomici (cellule muscolari lisce, apparato circolatorio, sistemi di regolazione fisiologica) danno luogo a manifestazioni che si instaurano rapidamente (per es. broncospasmo nell'asma allergico o shock circolatorio nel quadro di tipo anafilattico).
Quando, in altre circostanze o grazie ad altri antigeni (come virus o batteri), si ha la stimolazione dei linfociti TH1, si osservano meccanismi immunopatogenetici causati da cellule e clinicamente si hanno reazioni di ipersensibilità ritardata (come nel caso della tubercolosi).
In generale il coinvolgimento dei macrofagi e delle cellule NK (Natural Killer) nei confronti di particolari agenti patogeni induce la sintesi di citochine (IL-12, da parte dei macrofagi; IL-2 e IFN-γ da parte dei linfociti), che attivano la partecipazione di numerose componenti cellulari, il cui scopo consiste nel contenimento dell'aggressione e nell'eliminazione finale dell'agente patogeno. In tale contesto, se i linfociti T vengono sottoposti a stimolazione continua e se vengono prodotte citochine per tempi abnormi, si osserva un danno secondario dei tessuti vicino ai quali si svolgono le reazioni o può instaurarsi una forma di contenimento ben conosciuta come reazione di tipo granulomatoso. Se questo tipo di risposta con trend TH1 ha un significato ben preciso nei confronti di particolari aggressori (parassiti e batteri endocellulari, verso i quali gli anticorpi non sono adeguatamente efficaci), tuttavia essa può verificarsi anche nei confronti di alcuni antigeni che causano fenomeni di ipersensibilità ritardata come la dermatite da contatto e che sono la conseguenza di un alterato controllo della risposta immunitaria. Infatti si tratta di particolari molecole, solitamente di esigue dimensioni, che vengono associate a cellule presentanti l'antigene (per es. le cellule di Langherans sulla cute) che inducono un trend di risposta TH1 mediato, coinvolgente in forma locale o sistemica l'afflusso di cellule (macrofagi) che liberano sostanze litiche responsabili della sintomatologia osservata in questi casi.
Patologie autoimmuni
Nell'ambito delle malattie autoimmuni (siano esse sistemiche od organo-specifiche) i numerosi progressi conseguiti hanno riguardato sia una migliore codificazione delle patologie note, sia un ampliamento di conoscenze relative a nuove forme morbose (per es. l'inserimento fra le malattie autoimmuni della vitiligine e dell'orticaria cronica idiopatica da autoanticorpi). In studi su particolari ceppi di topo sono stati identificati gruppi di geni non MHC-correlati (MHC, Major Histocompatibility Complex), che rappresentano il substrato condizionante per l'espressione fenotipica delle malattie autoimmuni.
Di particolare interesse sono state le analisi sul ruolo della tolleranza centrale e sulla teoria della selezione clonale. Infatti, a livello del timo i 9/10 della popolazione linfocitaria che stabilisce un legame altamente affine con frazioni proteiche self vengono eliminati o inattivati. È grazie a questo meccanismo che in condizioni fisiologiche risultano bloccati i cloni autoreattivi. Un processo noto come apoptosi regola le fasi di autoeliminazione cellulare; molti studi si sono concentrati su particolari molecole in grado di attivare i processi apoptotici. Un ruolo rilevante è operato dal sistema Fas/Fas-L: se per alterazioni genetiche o altre cause il segnale di attivazione apoptotica viene meno, in alcuni ceppi di topo si genera una malattia caratterizzata da abnorme crescita linfoproliferativa, con genesi di molecole autoanticorpali e danno renale (glomerulonefrite).
Osservazioni sperimentali in malattie autoimmuni organo-specifiche hanno messo in evidenza come numerosi bersagli dell'aggressione umorale (autoanticorpi) e/o cellulo-mediata siano enzimi (per es. perossidasi tiroidea nelle tireopatie autoimmuni; tirosinasi nella vitiligine; H+K+adenosintrifosfatasi nella gastrite autoimmune/anemia perniciosa), e come la perdita di tolleranza sia correlata a uno o più epitopi presenti nelle molecole autoantigeniche proprie di un determinato tessuto. Malgrado l'esistenza di malattie organo-specifiche che hanno diversa espressione clinica, oggi si tende a stabilire un'interpretazione patogenetica del danno su base unitaria. Il primo passo consiste nell'attivazione di linfociti T potenzialmente autoreattivi. Questi cloni sono naturalmente presenti nell'organismo in quantità esigue, però non sono del tutto assenti; in fase quiescente all'interno del sistema linfatico non provocano danno in condizioni fisiologiche. Se le APC si combinano con un autoantigene, esse lo possono esporre ai cloni T autoreattivi. In generale una presentazione efficiente dell'autoantigene è resa possibile da cellule APC professioniste: in tal modo i linfociti T, grazie anche all'intervento di citochine, possono proliferare e volgere l'espansione clonale in senso TH1. Sembra che una forte espansione dei linfociti TH1 sia all'origine del salto di qualità che consente a una 'normale' reazione autoimmunitaria di trasformarsi in evento patologico istolesivo (malattia autoimmune). I linfociti T così attivati tendono a mantenere alto il proprio livello di replicazione, con un'amplificazione in situ favorita dal riconoscimento dell'antigene, senza che ci sia ulteriore necessità di segnali accessori. Ne consegue, inoltre, un insieme di fenomeni che comporta un'azione citotossica da parte di linfociti T CD8+ che esplicano autoreattività citotossica sulle cellule dell'organo bersaglio. D'altro canto una funzione helper che stimoli cloni B autoreattivi ad andare incontro a proliferazione comporta un meccanismo parallelo di differenziazione cellulare con genesi di autoanticorpi potenzialmente patogeni.
Tra gli esempi di patologia autoimmune a carattere sistemico, nel cui ambito sono stati compiuti progressi notevoli per la comprensione della patogenesi, un rilievo particolare ha assunto la sindrome da anticorpi antifosfolipidi.
Questa sindrome, della quale si distinguono una forma primaria e una secondaria associata a LES (Lupus Eritematoso Sistemico) o ad altre connettiviti, è caratterizzata da manifestazioni trombotiche artero-venose recidivanti, piastrinopenia, aborti ricorrenti, e presenza di autoanticorpi il cui bersaglio sono molecole di fosfolipidi. Sono colpite prevalentemente le femmine nel rapporto 2:1 rispetto al sesso maschile.
Nella forma primitiva sembra significativa la correlazione con l'allele DQB1 *0301 del sistema HLA, equivalente nell'uomo al sistema MHC del topo. Il carattere fondamentale della lesione istopatologica è correlato all'azione degli autoanticorpi antifosfolipidi. Essi rappresentano un gruppo piuttosto eterogeneo di molecole che agiscono verso strutture anioniche di fosfolipidi. Un anticorpo ben noto è il LAC (Lupus Anti-Coagulant). Si tratta di un anticorpo di classe IgG o IgM che genera, se presente, un allungamento del tempo di coagulazione nei test fosfolipido-dipendenti. Il LAC agisce nei confronti del cosiddetto complesso attivatore della protrombina. Le conseguenze di un anticoagulante, invece di essere di tipo emorragico, sono, nel caso della sindrome da anticorpi antifosfolipidi, di carattere ipercoagulativo trombigeno. Probabilmente il LAC, agendo con la molecola di trombomodulina e di alcuni fosfolipidi, blocca l'azione della proteina C (attivata per l'appunto dal sistema trombina-trombomodulina); l'inattivazione della proteina C si accompagna a un processo che favorisce la coagulazione e diminuzione della fibrinolisi.
Altre osservazioni hanno dimostrato che anticorpi antifosfolipidi diversi dal LAC possono essere presenti nel sangue, in grado di riconoscere epitopi appartenenti a diversi fosfolipidi. In particolare, sembra che l'azione verso una molecola nota come b2-glicoproteina I (b2-GPI) induca una riduzione o un blocco delle funzioni anticoagulanti da questa esplicata, con l'instaurarsi di un processo di attivazione trombinico. Nel quadro clinico ha particolare rilievo il coinvolgimento del sistema nervoso centrale.
Ai fini diagnostici un punto importante concerne la differenziazione tra forme primitive e il quadro secondario a LES o ad altre malattie del connettivo. Criteri internazionali di diagnosi sono stati proposti e prevedono criteri maggiori (trombosi arteriose e venose, aborti ricorrenti, piastrinopenia, presenza di LAC e di anticorpi anticardiolipina) e minori (cefalea, ipertensione polmonare, caratteristica livedo reticularis). La terapia si basa sull'impiego di anticoagulanti e/o antiaggreganti, il ricorso alla plasmaferesi e all'impiego di immunosoppressori.
bibliografia
C. Cunningham-Rundles, Disorders of the IgA system, in Immunologic disorders in infants and children, ed. E.R. Stiehm, Philadelphia 1973, 1996⁴, pp. 423-42.
Fundamental immunology, ed. W.E. Paul, New York 1984, 1993³.
F. Aiuti, G. Luzi, Deficienze immunologiche congenite e acquisite, in Trattato italiano di medicina interna, a cura di M. Negri, Firenze 1987.
S. Romagnani, F. Caligaris Cappio, Immunologia clinica e allergologia, Torino 1991.
M.E. Conley, M. Larche, V.R. Bonagura et al., Hyper IgM syndrome associated with defective CD40-mediated B cell activation, in Journal of clinical investigation, 1993, 94, pp. 1404-09.
L. Steinman, Autoimmune disease, in Scientific American, 1993, 269, pp. 106-14.
P. Macchi, A. Villa, S. Giliani et al., Mutations of JAK3 gene in patients with autosomal severe combined immunodeficiency (SCID), in Nature, 1995, 377, pp. 65-68.
D.P. Huston, The biology of the immune system, in The journal of the American medical association, 1997, 278, pp. 1804-14.
J.M. Puck, Primary immunodeficiency diseases, in The journal of the American medical association, 1997, 278, pp. 1835-41.
J.M. Puck, A.E. Pepper, P.S. Henthorn et al., Mutation analysis of IL2RG in human X-linked severe combined immunodeficiency, in Blood, 1997, 89, pp. 1968-77.
L.J. Rosenwasser, Interleukin-4 and the genetics of atopy, in New England journal of medicine, 1997, 337, pp. 1766-68.
K. Saar, K.H. Chrzanowska, M. Stumm et al., The gene for ataxia-telangiectasia variant, Nijmegen breakage syndrome, maps to a 1-cM interval on chromosome 8q21, in American journal of human genetics, 1997, 60, pp. 605-10.
IUIS Scientific Group, Primary immunodeficiency diseases, in Clinical experimental immunology, s. 1, 1999, 118, pp. 1-28.