
Femtochimica
Femtochimica
Il termine, composto da femtosecondo (10−15 s) e chimica, ha avuto origine nell'ultimo ventennio del 20° sec., all'interno del gruppo di ricerca diretto dal premio Nobel A. Zewail al Caltech (California Institute of Technology), per designare quella branca della cinetica chimica che studia i processi elementari ultraveloci, che avvengono cioè in tempi brevissimi, dal miliardesimo al milionesimo di miliardesimo di secondo. La scala dei tempi in ore, minuti e secondi, fino ai millisecondi, del mondo macroscopico, è inadeguata per descrivere il movimento di oggetti del mondo microscopico. Seguire in tempo reale la rottura e la formazione dei legami chimici in una reazione, è indispensabile per individuare i percorsi che i reagenti compiono trasformandosi nei prodotti, ossia per caratterizzare tutti gli stadi intermedi che portano dai reagenti iniziali ai prodotti finali.
Per comprendere le ragioni per cui i tempi necessari per seguire i movimenti atomici o molecolari sono completamente al di là della scala dei tempi del mondo macroscopico, è sufficiente considerare brevemente le dimensioni e i movimenti degli atomi e delle molecole: a) i legami chimici tra atomi nelle molecole hanno dimensioni di 0,1 nm e le distanze tra molecole sono dello stesso ordine di grandezza, ossia miliardi di volte più piccole di quelle tra oggetti del mondo macroscopico; b) le molecole in un liquido o in un gas si muovono a velocità elevatissime, dell'ordine di 104-105 cm/s; c) i legami degli atomi nelle molecole dei reagenti si rompono e nuovi se ne formano per dare luogo ai prodotti della reazione. I processi sono rapidissimi e avvengono in tempi dell'ordine di frazioni di picosecondo. I tempi necessari per attraversare le distanze interatomiche sono dell'ordine delle centinaia di femtosecondi.
La branca della chimica che si occupa della velocità e dei meccanismi delle reazioni prende il nome di cinetica chimica. Lo studio tanto della cinetica quanto dei meccanismi di reazione è stato al centro dell'attenzione dei chimici per quasi due secoli. Per lungo tempo, misure nel dominio dei tempi veloci hanno rappresentato un problema insolubile per la mancanza di strumenti adatti. Con la comparsa dei laser con impulsi di durata inferiore ai tempi di rottura dei legami chimici queste misure sono diventate possibili.
I laser pulsati a rubino, con tempi di nanosecondi, sono comparsi nel 1961. Impulsi più brevi sono stati realizzati nel 1966 con i laser a coloranti e nel 1990 con il laser a titanio-zaffiro in strutture a bloccaggio di modo (mode-locking): si sono ottenuti così impulsi di picosecondi e poi di femtosecondi.
Le grandi possibilità offerte dai laser pulsati hanno permesso di estendere ai tempi ultracorti la spettroscopia risolta nel tempo, ossia le tecniche chimico-fisiche che permettono di seguire la variazione nel tempo dell'interazione radiazione-materia. Il processo primario più semplice di interazioneesistente tra una molecola e la radiazione elettromagnetica è l'assorbimento di fotoni (fotoeccitazione) che consente alla molecola di passare dallo stato di energia più basso, che è quello fondamentale, a stati vibroelettronici eccitati di energia maggiore. In uno stato eccitato la molecola si trova fuori equilibrio e cerca di smaltire l'eccesso di energia per raggiungere una situazione stabile. I processi che guidano il sistema verso l'equilibrio vengono detti di rilassamento dell'energia e avvengono su scale temporali estremamente brevi, dell'ordine dei picosecondi o dei femtosecondi. Il modo più semplice per una molecola per ritornare allo stato fondamentale sarebbe l'emissione di uno o più fotoni. La maggior parte dei processi di rilassamento dell'energia avviene però senza emissione di radiazione, in quanto tali processi non radiativi sono normalmente molto più veloci dell'emissione spontanea di fotoni da stati eccitati. Conseguentemente, la molecola eccitata si riassesta rapidamente con processi non radiativi, trasferendo l'energia dal livello vibrazionale eccitato a livelli inferiori fino al raggiungimento del livello vibrazionale più basso del livello elettronico eccitato (rilassamento vibrazionale). Lo stato raggiunto a causa dei processi di rilassamento è anch'esso metastabile. Esso può decadere sia per emissione di un fotone (fluorescenza), sia per ulteriori processi di rilassamento non radiativo, sia per trasferimento di energia a stati di tripletto (intersystem crossing). La vita media dello stato è ovviamente determinata dal processo di rilassamento più veloce.
Per realizzare esperimenti in tempi rapidissimi è necessario conoscere con grande esattezza il tempo zero, ossia l'istante in cui il processo inizia. Questa condizione può essere facilmente realizzata innescando la reazione per interazione fra la molecola in esame e un impulso laser ultracorto. Per questa ragione, la maggior parte delle reazioni studiate con l'intento di caratterizzare lo stato di transizione sono reazioni fotochimiche. È però importante che gli eventi successivi alla eccitazione che inizia la catena dei processi reattivi siano più veloci della reazione stessa. Se la successione di questi processi contiene uno stadio lento, come, per es., uno stadio di diffusione dei reagenti, ciò che si misura è l'effetto di questo stadio lento e sarà quindi altamente improbabile caratterizzare l'evento reattivo.
La misura dei tempi ultraveloci
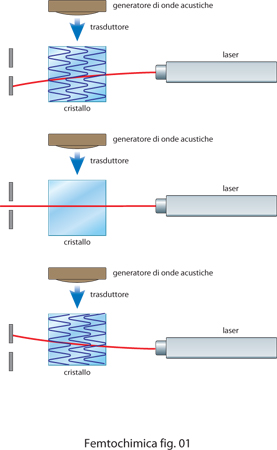
I laser pulsati più utilizzati sono quelli a stato solido, in particolare con il mezzo attivo costituito da una barretta di zaffiro con impurezze di titanio. All'interno di una cavità laser si creano molti modi di oscillazione del campo elettromagnetico. Per forzare la cavità a emettere impulsi di radiazione con tutti i modi in fase si può fare ricorso a un sistema, del tipo rappresentato in fig. 1, che utilizza un modulatore acustoottico.
Il principio di funzionamento è molto semplice: un'onda acustica generata in un cristallo, produce l'equivalente di un reticolo di diffrazione che devia il raggio laser che l'attraversa. Quando l'onda acustica è presente il raggio è deviato, mentre quando l'onda acustica è assente il raggio si propaga normalmente. Modulando quindi l'ampiezza dell'onda acustica con un'adatta frequenza, che dipende dall'inverso della lunghezza della cavità, si forzano i modi della cavità a guadagnare tutti nello stesso istante e quindi a essere in fase. Nel 1991 è stato scoperto che i laser a titanio-zaffiro danno luogo a impulsi a femtosecondi con un meccanismo dell'agganciamento di fase tra modi diversi che avviene automaticamente a causa della variazione dell'indice di rifrazione della barretta, che è dovuta all'effetto Kerr. La barretta funziona alternativamente da lente divergente o da lente convergente, modulando di conseguenzal'intensità all'interno della cavità laser.
I laser a titanio-zaffiro hanno impulsi accordabili in frequenza dal rosso, vale a dire da circa 750 nm, fino al vicino infrarosso (∼900 nm). L'accordabilità in frequenza può essere estesa fino all'infrarosso medio (∼3500 nm) usando amplificatori parametrici, capaci di generare, per mezzo di processi non lineari, due frequenze diverse, tali che la loro somma sia eguale alla frequenza di eccitazione del laser. Per generazione di frequenze somma in cristalli non lineari, si può poi estendere l'accordabilità nella direzione delle alte frequenze fino al vicino ultravioletto (∝200 nm).
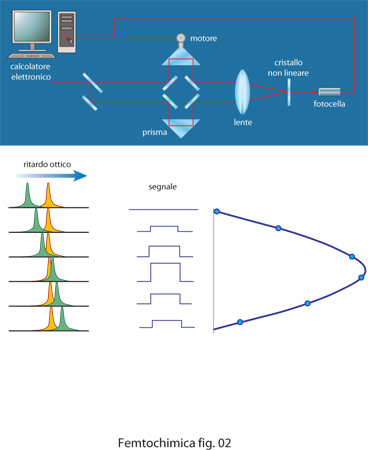
Il singolo impulso laser ultracorto a femtosecondo, può essere amplificato fino a energie di alcuni mJ, corrispondenti a una potenza istantanea elevatissima, dell'ordine di molti GW. Se gli impulsi vengono poi focalizzati con una lente, si possono raggiungere potenze di TW superiori alla soglia di danneggiamento di quasi tutti i materiali di cui sono fatte le componenti ottiche (specchi, reticoli, lenti) di un laser. Per amplificare gli impulsi a femtosecondi è quindi necessario prima allungarli temporalmente fino a centinaia di picosecondi in modo da ridurre la potenza istantanea, quindi amplificarli e infine comprimerli di nuovo. Al fine di ottenere la misura viene utilizzata la proprietà di molti cristalli, denominati non lineari, di sommare insieme l'energia di due fotoni per produrre un fotone di energia doppia. Lo schema di una misura di autocorrelazione è rappresentato in fig. 2. Il raggio laser viene diviso in due usando uno specchio semitrasparente e ai due raggi così ottenuti viene fatto percorrere un cammino ottico differente prima di ricongiungerli insieme focalizzandoli su un cristallo non lineare. Se i due impulsi arrivano sul cristallo a tempi diversi la loro sovrapposizione è nulla e, quindi, non si ha emissione apprezzabile di fotoni di frequenza doppia. Spostando in maniera continua il prisma nel percorso di uno dei due raggi si può avvicinare uno dei due impulsi all'altro, fino a sovrapporli e infine a invertire il tempo con cui arrivano sul cristallo non lineare. Quanto più i due impulsi sono sovrapposti, tanto maggiore è il numero di fotoni che arrivano nello stesso istante sul cristallo e dunque tanto maggiore è il numero di fotoni di frequenza doppia emessi. Misurando quindi l'intensità della radiazione emessa si ricostruisce la forma temporale dell'impulso.
La durata di impulsi laser di picosecondi o di alcune centinaia di femtosecondi, è anche misurabile utilizzando una streak camera. Il principio di funzionamento di tale strumento è la conversione di fotoni in elettroni che, una volta creati, attraversano un campo elettrico variabile rapidamente nel tempo. Due fotoni che arrivano con un ritardo Δt creano, quindi, due elettroni anch'essi con un ritardo Δt. Gli elettroni passando nel campo elettrico a tempi diversi subiscono una differente deflessione e vengono quindi separati spazialmente. Quando si arriva però a tempi di pochi femtosecondi o ancora più brevi, sia le misure di autocorrelazione sia quelle con la streak camera diventano molto difficili e complesse. In questo caso conviene calcolare il tempo dall'ampiezza a mezza altezza della banda di frequenze dell'impulso, utilizzando la nota relazione del principio di indeterminazione.
Tecniche di spettroscopia ultraveloce
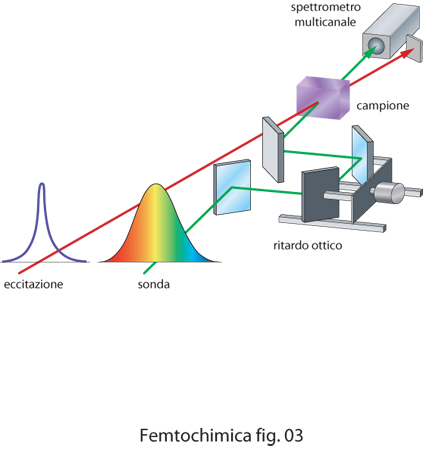
La maggior parte dei metodi spettroscopici risolti nel tempo utilizzati in f. è basata sulla tecnica detta di eccitazione e sonda o anche di pompa e prova (pump and probe), ossia sull'uso di due impulsi ultracorti, uno che serve per eccitare il campione (impulso di eccitazione) e l'altro, inviato sul campione con un ritardo variabile (impulso di sonda), per identificare le specie transienti o i processi di eccitazione molecolare creati dal primo impulso. Tra le tecniche spettroscopiche utilizzate in f., particolare rilevanza riveste l'assorbimento transiente illustrato schematicamente in fig. 3. L'assorbimento transiente è largamente utilizzato per caratterizzare specie chimiche a vita molto breve o per seguire l'evoluzione temporale di stati eccitati di una molecola. In un esperimento di assorbimento transiente un impulso ultracorto molto intenso di pompa è utilizzato per eccitare la molecola dallo stato fondamentale a uno stato di energia più alta, che può rapidamente evolvere sia dissociando la molecola in radicali o specie ioniche, sia trasferendo l'energia ad altri stati molecolari. Un impulso di sonda viene quindi inviato sul campione con un ritardo calcolato. Il ritardo ottico si realizza facendo attraversare a uno dei due raggi un cammino variabile semplicemente spostando, con l'aiuto di un motore comandato da un calcolatore, due specchi o un prisma. La radiazione usata per l'impulso di sonda può essere sia bianca, ossia radiazione che si estende su quasi tutto lo spettro visibile, sia radiazione centrata a una frequenza diversa da quella del raggio di pompa, ma tale da essere assorbita dalle nuove specie presenti. L'impulso di sonda è utilizzato per ottenere due spettri di assorbimento del campione, uno in presenza e uno in assenza del raggio di pompa. La differenza tra i due spettri fornisce lo spettro della specie transiente. Utilizzando ritardi variabili si ottiene lo spettro risolto nel tempo.
Anche la LIF (Laser-induced fluorescence, fluorescenza indotta da laser) è molto utilizzata in misure di femtochimica. Il grande vantaggio di questa tecnica è il bassissimo rumore di fondo rispetto alle misure di assorbimento. In questo caso la fluorescenza emessa dal campione a seguito dell'assorbimento di un impulso ultracorto è registrata in funzione del tempo oppure utilizzando una streak camera o, con maggiore risoluzione temporale, usando un secondo impulso laser per attivare il sistema di misura.
Tutte le misure del tipo pump and probe richiedono l'agganciamento di fase dei due impulsi laser e una corretta eliminazione di tutti i contributi incoerenti che disturbano la misura. Per questa ragione, spesso si preferiscono tecniche che combinano la coerenza di più raggi laser. Le tecniche di ottica non lineare di mescolamento a quattro onde, che utilizzano l'interazione di tre campi elettrici non lineari per produrne un quarto, sono fortemente preferite da questo punto di vista. Tra queste sono particolarmente utilizzate quella dei reticoli transienti e quella dell'eco a tre fotoni.
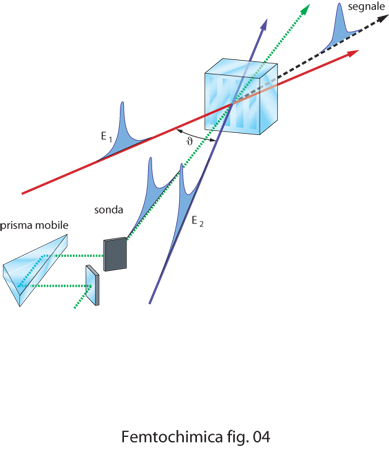
La tecnica dei reticoli transienti, schematizzata in fig. 4, è basata sull'uso di due impulsi laser di eccitazione E1 ed E2 che per interferenza creano nel campione un reticolo di diffrazione transiente di fase. La spaziatura del reticolo è regolata dall'angolo ϑ tra le direzioni dei due raggi di eccitazione incidenti. L'impulso di sonda, inviato con ritardi variabili, viene diffratto dal reticolo generando un segnale che varia nel tempo in funzione del ritardo ottico, con intensità che dipende dalla vita media degli stati elettronici eccitati dagli impulsi.
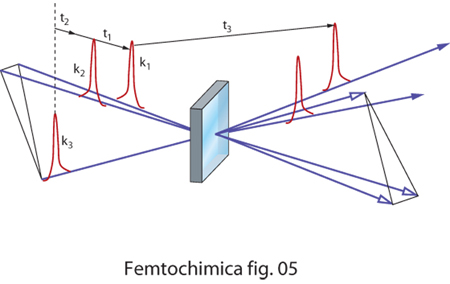
Anche nel caso relativo all'eco di fotoni (fig. 5), vengono utilizzati tre impulsi. I primi due impulsi creano una popolazione di stati eccitati in fase tra di loro. Con il passare del tempo la memoria della fase tende a perdersi e viene recuperata dal terzo impulso. Come in un normale effetto di eco l'intensità del segnale è proporzionale al ritardo tra il terzo impulso e la coppia di impulsi di eccitazione e decade con il tempo naturale di decadimento degli stati eccitati, indipendentemente dalla presenza di effetti di inomogeneità del sistema molecolare.
Per misure su molecole in fase gassosa conviene associare la spettroscopia risolta nel tempo alla tecnica dei raggi molecolari supersonici che permette di ottenere molecole completamente isolate nel vuoto a temperature molto basse, dell'ordine di qualche K. In queste condizioni i livelli vibrazionali eccitati delle molecole, normalmente popolati a temperatura ambiente, sono completamente spopolati. Utilizzando raggi molecolari, sono stati studiati i processi di decomposizione, al livello microscopico, di molte molecole biatomiche e triatomiche e perfino di molecole poliatomiche complesse, nonché molte reazioni di scambio tra atomi e molecole. Altrettanto importante è l'associazione della spettroscopia laser ultraveloce alla spettroscopia di massa. In questo caso gli impulsi laser di pompa producono la dissociazione delle molecole e la spettroscopia di massa è utilizzata per caratterizzare i frammenti formatisi, spesso ioni o radicali.
Meccanismi elementari delle reazioni chimiche
Dalla fine degli anni Ottanta del 20° sec., con la comparsa di laser a stato solido e con lo sviluppo degli amplificatori parametrici, che permettono di amplificare la radiazione accordandola su un vasto campo di frequenze, si è aperta una nuova era per lo studio dei meccanismi delle reazioni chimiche e della dinamica molecolare. Intere classi di reazioni sia intramolecolari sia intermolecolari, per le quali era stato finora impossibile individuare i cammini di reazione perché estremamente veloci, sono state ora chiarite grazie alle tecniche di spettroscopia ultraveloce illustrate precedentemente. Risultati importanti sono stati ottenuti nello studio dei processi di dissociazione di molecole semplici, per le quali è stato completamente caratterizzato lo stato di transizione. Per una reazione elementare del tipo A+BC → [ABC]ǂ → AB+C, lo stato di transizione [ABC]╪ è definito come l'insieme di tutte le possibili famiglie di configurazioni di ABC attraverso le quali le particelle reagenti evolvono per dar luogo ai prodotti. Lo stato di transizione è normalmente rappresentato semplificando la superficie multidimensionale dell'energia potenziale del sistema al caso bidimensionale, nel quale compare un solo percorso di reazione, rappresentativo di tutti quelli possibili. Lo stato di transizione idealizzato in questo modo è situato alla sommità della barriera di potenziale che separa i reagenti dai prodotti.
Riguardo alla vasta mole di ricerche accumulatesi sui meccanismi di reazione studiati con tecniche di spettroscopia risolta nel tempo, ci si limiterà a descrivere alcune problematiche di carattere generale che offrono una panoramica molto chiara delle linee fondamentali di ricerca della f. e dei contributi che sono stati apportati alla conoscenza dei meccanismi di reazione.
Un esempio di reazioni velocissime, che si presta perfettamente a illustrare in quale modo è articolata e come procede una misura di spettroscopia risolta nel tempo, è quello della dissociazione della molecola di I−CN secondo la reazione
I−CN → [ I····CN]╪* → I+CN
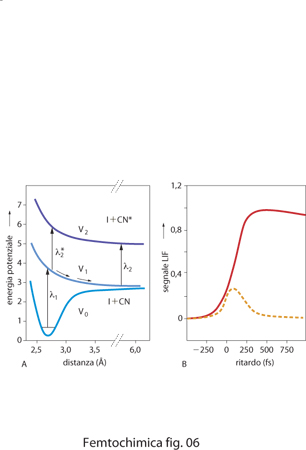
dove l'apice ╪ indica lo stato di transizione e l'asterisco * designa un livello eccitato. Nella fig. 6 A sono indicate le curve di energia potenziale dello stato fondamentale V0, del primo stato eccitato repulsivo V1 e di un secondo stato eccitato repulsivo V2 della molecola I−CN. Un fotone di lunghezza d'onda λ1=307 nm dell'impulso di pompa eccita la molecola nello stato V1, definendo l'istante iniziale. L'impulso laser di sonda a λ2=388,5 nm, corrispondente al massimo di assorbimento del frammento libero CN, è quindi inviato sulla molecola a intervalli di tempo variabili, ritardati rispetto all'impulso di pompa. I risultati delle misure di fluorescenza sono poi riportati in funzione del ritardo temporale tra l'impulso di pompa e quello di sonda (fig. 6 B). Al tempo zero e fino a quando il ritardo tra i due impulsi è breve, il gruppo CN fa ancora parte del complesso [ I····CN]╪*, non avendo avuto il tempo di separarsi completamente dall'atomo di iodio spostandosi lungo la curva di potenziale V1. In queste condizioni non si osserva fluorescenza perché il complesso non può assorbire il fotone λ2. Quando invece il ritardo arriva a circa 250 fs, sufficiente per permettere al gruppo CN di giungere a una distanza R di separazione dall'atomo di iodio superiore a circa 0,6 nm, comincia la fluorescenza perché ora il gruppo CN libero assorbe esattamente alla frequenza del fotone λ2, come mostrato dalla curva a tratto pieno. Per stimolare l'emissione fluorescente a tempi minori, è necessario invece usare un impulso di sonda accordato a frequenza leggermente diversa λ2*, che possa essere assorbita dal complesso [I····CN]╪*. Il complesso fluoresce finché il gruppo CN non si è allontanato e a questo punto la fluorescenza si interrompe (curva tratteggiata in fig. 6 B).
Un esempio di caratterizzazione dello stato di transizione in una molecola poliatomica è la reazione di fotodissociazione dell'azometano. La reazione è conosciuta da lungo tempo e in vari laboratori si era cercato con metodi indiretti di stabilire il meccanismo col quale l'azometano si dissocia per dare origine a due radicali metili e a una molecola di azoto. Prima dell'introduzione di sorgenti laser a impulsi ultracorti, non era stato possibile stabilire con certezza se la dissociazione avvenisse attraverso un unico meccanismo concertato
CH3−N=N−CH3→CH3 ∙+CH3 ∙+N2
oppure in due fasi successive secondo lo schema
CH3−N=N−CH3→CH3−N2∙+CH3∙ →CH3∙+CH3∙N2
dove il segno ∙ indica la presenza di un radicale, ossia di una molecola con un elettrone spaiato. Nel primo caso i due legami sono rotti contemporaneamente, mentre nel secondo la reazione produce un radicale CH3−N2 ∙ il quale solo successivamente libera una molecola di azoto. La formazione dell'intermedio CH3−N2∙ è stata dimostrata abbinando la spettroscopia di massa alle tecniche laser ultraveloci. L'esperimento mostra inequivocabilmente che i due eventi successivi di rottura dei legami C−N avvengono su scale temporali estremamente brevi, ma comunque distinguibili. Utilizzando impulsi di 80 fs e monitorando la comparsa della massa relativa all'intermedio CH3−N2 ∙ si è visto che questo si forma in circa 70 fs e decade con una costante di tempo di circa 100 fs. La molecola di azoto e gli altri frammenti compaiono con una costante di tempo di circa 100 fs. Questi risultati hanno permesso di dimostrare che il meccanismo è di tipo consecutivo e non concertato. L'esperimento mette in luce, inoltre, che il processo di dissociazione avviene con tempi paragonabili a quelli di un ciclo vibrazionale.
Una classe di reazioni intramolecolari molto importante è quella dell'isomerizzazione. Tra queste l'isomerizzazione cis-trans è una delle più semplici reazioni intramolecolari. Come esempio consideriamo la molecola di azabenzene, C6H5−N=N−C6H5 costituita da due anelli benzenici legati a un doppio legame N=N, che può esistere in forma trans o in forma cis. La forma trans ha una struttura planare con i due anelli benzenici giacenti da parti opposte rispetto all'asse passante per il doppio legame N=N. La forma cis è anch'essa planare, ma i due anelli benzenici giacciono dalla stessa parte. La trasformazione di una forma nell'altra può avvenire sia per via termica, sia per fotoeccitazione e comporta una variazione consistente della distanza tra i due anelli. Il processo di isomerizzazione trans-cis avviene attraverso due meccanismi completamente diversi, a seconda della lunghezza d'onda utilizzata nella fotoeccitazione. Se per assorbimento di un fotone si raggiunge il più basso stato elettronico eccitato, la conversione dalla forma trans a quella cis avviene per rotazione nel piano molecolare (inversione). Nello stadio iniziale del processo uno degli anelli benzenici ruota nel piano allineandosi lungo l'asse N=N in circa 2,5 ps. Da questa posizione la molecola decade poi nuovamente nello stato fondamentale sia in configurazione cis sia in configurazione trans. Se invece si assorbe un fotone di frequenza maggiore che permette di raggiungere il secondo stato eccitato, la conversione si attua per rotazione fuori del piano di una metà della molecola attorno all'asse N=N. Il secondo stato eccitato decade in circa 200 fs in un minimo relativo, per rotazione di circa 20° di un anello benzenico perpendicolarmente al piano molecolare. Successivamente la molecola rilassa l'eccesso di energia vibrazionale raggiungendo un minimo relativo del primo stato eccitato dal quale decade poi nello stato fondamentale in circa 15 ps.
Reazioni molto veloci riguardano il trasferimento di protoni sia all'interno di molecole (trasferimento intramolecolare), sia tra molecole diverse, in particolare tra molecole di soluto e di solvente (trasferimento intermolecolare). Il processo più noto di tipo intermolecolare è la dissociazione degli acidi in acqua, secondo la ben nota reazione
HX+H2O → X−+H3O+
di importanza fondamentale in chimica e biochimica, che avviene in tempi dell'ordine del picosecondo. Simulazioni al calcolatore hanno dimostrato che l'effetto del solvente è quello di distorcere il potenziale lungo la coordinata di trasferimento del protone. In questo modo la funzione potenziale che ha un minimo quando l'idrogeno è collegato all'atomo X, progressivamente si deforma presentando prima due minimi simmetrici e poi trasferendo il minimo nella posizione in cui l'idrogeno è attaccato a una molecola di acqua. Il livello fondamentale di energia vibrazionale del protone è superiore alla debole barriera che separa i due minimi equivalenti nella situazione intermedia e pertanto non è necessario alcun effetto tunnel per trasferire il protone.
Una classe di reazioni intramolecolari semplici è rappresentata dal trasferimento protonico in stati elettronici eccitati. Molte delle molecole organiche che contengono gruppi ossidrilici, come per es. gli alcoli, si comportano da acidi forti nello stato eccitato, nonostante nello stato fondamentale siano soltanto acidi molto deboli, manifestando unicamente una debole tendenza a liberare protoni. Un esempio di molecole del tipo preso in esame, peraltro classico, può essere rappresentato dal fenolo C6H5−OH, che nello stato fondamentale dà origine allo ione fenato C6H5−O− solotanto in presenza di una base molto forte, mentre nello stato eccitato si comporta da acido forte.
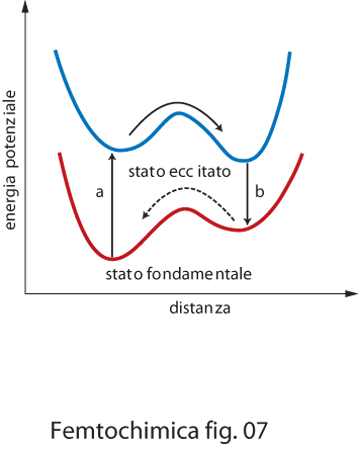
Qualora si produca la transizione allo stato eccitato in una molecola in cui sono presenti contemporaneamente un ossidrile e un gruppo accettore di protone, come un azoto o un altro atomo di ossigeno, il protone tende a migrare dal gruppo ossidrile, divenuto ora un acido forte, per attaccarsi all'atomo di azoto o di ossigeno. Tale reazione è tanto più rapida se il protone si trova già in prossimità del sito accettore. Nella fig. 7 è rappresentata la superficie di potenziale, nello stato fondamentale e in quello eccitato, lungo la coordinata di trasferimento del protone per una molecola di questo tipo contenente, per es., un legame OH e un atomo di azoto. Poiché il minimo di energia delle due curve non coincide, per assorbimento di un fotone (processo a), la molecola si viene a trovare in una situazione di instabilità. In questa condizione il meccanismo di trasferimento protonico è favorito e il sistema raggiunge un nuovo minimo corrispondente alla formazione del legame N−H. Il tempo di vita di questa specie è generalmente abbastanza lungo, dell'ordine di alcuni nanosecondi. Successivamente la molecola decade nello stato fondamentale per emissione di un fotone (processo b). La situazione è a questo punto capovolta. La struttura è poco stabile e si riassesta con il trasferimento del protone verso il sito occupato prima del processo di fotoeccitazione. In particolari situazioni possono essere stabilizzate strutture intermedie.
La spettroscopia ultraveloce, inoltre, ha dato contributi decisivi alla comprensione dei meccanismi di trasferimento di elettroni che regolano la fotosintesi, ove si ha la conversione dell'energia solare in energia chimica attraverso una serie di complessi processi che trasferiscono l'energia dei fotoni assorbiti in una zona della molecola, l'antenna (Ant), in un'altra zona della macromolecola, il centro di reazione (RC). L'evento primario nella conversione dell'energia solare in energia chimica al centro di reazione è il trasferimento di elettroni attraverso la membrana fotosintetica alla quale sono collegati sia l'antenna, sia il centro di reazione.
Le due grandi classi di sistemi fotosintetici sono la classe delle piante (clorofilla) e la classe dei batteri fotosintetici. Tra questi ultimi quelli di gran lunga più studiati con tecniche di spettroscopia ultraveloce sono i batteri porpora.
Nelle clorofille esistono due centri di reazione, i fotosistemi PS1 (Photosystem 1) e PS11 (Photosystem 11) che lavorano in serie: il fotosistema PS11 ossida l'H2O formando O2, mentre il fotosistema PS1 riduce l'NADP+ (nicotinamide adenina dinucleotide fosfato+). I batteri porpora hanno invece un solo centro di reazione che non riesce a ossidare l'H2O. Il fotosistema PS1 è formato da tre complessi proteici e funziona sia come antenna sia come centro di reazione. Ogni monomero raggruppa un gran numero di componenti, tra cui ben 12 proteine, 96 clorofille-a e 22 carotenoidi. Solo 6 clorofille-a formano però il centro di reazione vero e proprio. Le altre costituiscono due strutture di antenne, la struttura di base (Ant1) e la struttura accessoria (Ant0) che assorbe a frequenze più alte. L'energia dei fotoni raccolti dalle antenne viene trasmessa in circa 40 ps al principale donatore di elettroni di tutto il sistema, una coppia di clorofille-a, P700, che a seguito dell'eccitazione luminosa, trasferisce un elettrone a una serie di accettori che formano due rami, A e B.
L'elettrone è trasferito in circa 29 ps al primo accettore di elettroni, una clorofilla che viene denominata A0, probabilmente localizzandosi in 9 ps in un accettore accessorio Aacc, non ancora ben identificato. Da A0 l'elettrone è trasferito a un secondo accettore A1 (fillochinone) in circa 40 ps. L'elettrone continua poi a discendere la scala di potenziali redox fino a un centro ferro-zolfo, detto Fx. Dal centro Fx l'elettrone passa poi nell'ordine ai centri ferro-zolfo FA e FB e infine da FB a una ferrodossina solubile FD che riduce l'NADP+ con l'aiuto di una riduttasi. Una volta realizzata la separazione di cariche, la reazione oscura, ossia la serie di reazioni che avvengono senza assorbimento di fotoni, procede molto più lentamente, dando alla fine origine alla produzione di NADPH e/o di ATP (adenosintrifosfato).
Il sistema fotosintetico dei batteri porpora è quello di gran lunga più studiato. Il sistema consiste di un centro di reazione e di due tipi di antenne, un'antenna centrale LH1 (Light Harvesting 1) che circonda il centro di reazione e una periferica LH2 (Light Harvesting 2). La struttura dell'antenna LH2, recentemente risolta, include due anelli concentrici di nove polipeptidi con struttura a α-elica, che collegano due anelli di pigmenti, il primo di 18 batterioclorofille (B850) e il secondo di 9 batterioclorofille (B800). La funzione fondamentale delle batterioclorofille B800 è di assorbire la radiazione tra 790 e 810 nm. L'energia resta intrappolata all'interno del sistema, passando da una all'altra delle batterioclorofille B800 in circa 1 ps e poi da queste viene trasferita, in circa 700 ps, alle batterioclorofille B850. L'energia si sposta quindi in circa 700 fs all'interno del sistema delle B850 e successivamente viene trasferita in circa 3 ps all'antenna LH1, formata da 32 polipeptidi organizzati in due anelli concentrici che racchiudono un anello di 32 batterioclorofille (B875) per il fatto che assorbono a circa 875 nm. All'interno del sistema delle batterioclorofille B875 il trasferimento di energia è estremamente rapido (≈80 fs).
bibliografia
Femtochemistry, ed. F.C. De Schryver, S. De Feyter, G. Schweitzer, with the Nobel lecture of A. Zewail, Weinheim-New York 2001.