
Farmacologia molecolare
Farmacologia molecolare
Nella seconda metà del Novecento l'estensione della farmacologia a campi di ricerca, quali la biochimica enzimatica, le comunicazioni molecolari inter- e intracellulari, l'organizzazione e la dinamica molecolare dei recettori e il controllo molecolare della trascrizione genica, ha determinato l'affermazione del principio che il meccanismo di interazione molecolare costituisce la base razionale per la progettazione di un farmaco. Ma le origini della teoria molecolare dell'azione dei farmaci risalgono agli inizi del XX sec., quando Paul Ehrlich, nelle sue ricerche sugli arsenobenzoli, ipotizzò l'esistenza di un'entità teorica su cui doveva fissarsi il farmaco per agire (corpora non agunt nisi fixata): è questo il primo abbozzo concettuale di 'recettore'. Nell'era degli enzimi ‒ gli anni Quaranta e Cinquanta ‒ emerse l'ipotesi che questi catalizzatori delle reazioni biologiche fossero i siti molecolari d'azione dei farmaci, ipotesi che si affermò definitivamente con la scoperta dei farmaci antimetaboliti utilizzati nel trattamento di alcune neoplasie. La teoria recettoriale dell'azione dei farmaci riceveva definitive conferme sul piano sperimentale con la riproduzione in vitro della reazione farmaco-recettore e con le dimostrazioni in vivo delle localizzazioni tissutali dei recettori dei farmaci, per concretizzarsi infine nei sorprendenti successi terapeutici ottenuti da farmaci progettati sul modello recettoriale, come gli antagonisti dei recettori istaminici H2 (cimetidina, famotidina, ranitidina, nizatidina) nella cura dell'ulcera gastroduodenale, o gli antagonisti del recettore AT1 dell'angiotensina II (sartani) nella terapia dell'ipertensione.
Nell'ultimo decennio del Novecento, la decodificazione del genoma con la conseguente identificazione della struttura primaria delle proteine, l'utilizzo della 'fabbrica cellulare' per far esprimere geni e biosintetizzare proteine, la possibilità di creare mutazioni sperimentali nel codice genetico per produrre nuove proteine funzionali, la generazione di animali transgenici, portatori di un nuovo gene o carenti di un particolare gene, sono alcuni dei potenti strumenti di ricerca offerti dalla biologia molecolare alla farmacologia. Promettenti esempi di applicazione delle nuove biotecnologie alla ricerca farmacologica sono la produzione e l'utilizzazione terapeutica di proteine umane ottenute mediante la tecnica del DNA ricombinante (biofarmaci o farmaci cd. 'biotecnologici'), la creazione di linee cellulari esprimenti recettori umani su cui studiare l'azione di farmaci noti o modellare nuovi farmaci, l'analisi intracellulare dei segnali molecolari attivati dal farmaco o la creazione, mediante manipolazioni del genoma, di modelli animali di malattie umane su cui studiare l'efficacia terapeutica dei farmaci. Il sequenziamento del genoma umano ha inoltre fornito alcune risposte ai quesiti posti dalla variabilità individuale di risposta ai farmaci. La possibilità di stratificare i pazienti sulla base del loro profilo genico ottenuto con la tecnica dei microarrays ha permesso in alcuni casi di associare l'efficacia terapeutica o la comparsa di effetti collaterali dei farmaci a un determinato genotipo. L'esistenza di un polimorfismo genetico nella popolazione ha fatto profilare all'orizzonte la possibilità di creare farmaci personalizzati, scelti e modellati in base ai dati dell'analisi genomica del singolo paziente.
Peptidi, proteine e biofarmaci
La scoperta che peptidi e proteine vengono utilizzati dall'organismo, oltre che come componenti strutturali della materia vivente, anche per trasmettere messaggi intercellulari su brevi distanze (paracrinia) rappresenta uno dei più rilevanti contributi alla conoscenza delle comunicazioni molecolari degli eucarioti. Caratteristica dei messaggeri peptidici è l'elevato grado di diversità e selettività molecolare: si conoscono attualmente più di cento diverse famiglie peptidiche, tutte riconosciute da selettivi recettori cellulari. Inoltre, lo stesso sistema peptidico (peptide-recettore) è spesso utilizzato nell'organismo per modulare differenti funzioni in diversi organi e apparati. Nel sistema nervoso centrale il messaggio peptidico si aggiunge e si sovrappone a quello sinaptico. La comunicazione extrasinaptica nel cervello è una delle acquisizioni più innovative dell'ultimo ventennio. L'ampia varietà e l'alta selettività dei messaggeri peptidici ben si adeguano alla necessità di informare selettivamente le numerose, diverse e spesso distanti popolazioni neuronali senza ricorrere a contatti sinaptici.
La scoperta del primo neuropeptide avvenne nel 1930, a opera del giovane studente Ulf S. von Euler. Alla sostanza P di von Euler si aggiunsero dopo circa quarant'anni altri peptidi dello stesso tipo e all'intera famiglia fu dato il nome generico di tachichinine o neurochinine. Le tachichinine (sostanza P, neurochinina A e neurochinina B) e i loro tre principali tipi di recettori (NK1, NK2, NK3) sono ampiamente distribuiti sia nel sistema nervoso centrale che in periferia. Questi peptidi eccitano neuroni, evocano risposte comportamentali, sono potenti vasodilatori e secretagoghi e fanno contrarre (direttamente o indirettamente attraverso la liberazione di trasmettitori sinaptici) la muscolatura liscia di molti organi. La sostanza P è coinvolta nella trasmissione del dolore e, insieme alle altre tachichinine, in fenomeni di broncocostrizione patologica (asma) e di infiammazione. Di recente è stata inoltre evidenziata la possibilità che la sostanza P agisca come modulatore delle risposte immunitarie e infiammatorie nel sistema nervoso centrale. I recettori delle tachichinine costituiscono importanti bersagli farmacologici per lo sviluppo di farmaci analgesici, antinfiammatori e immunomodulatori. Nel 1940 Eduardo Braun Menéndez, Irvin H. Page e Oscar M. Helmer, rispettivamente in Argentina e negli Stati Uniti, dimostrarono che l'azione enzimatica della renina, secreta dal rene, sulle proteine del plasma generava un peptide vasocostrittore e potentemente ipertensivo, l'angiotensina. Dieci anni dopo furono identificate due forme di angiotensina, denominate angiotensina I e angiotensina II, quest'ultima prodotta dalla prima a opera di un enzima denominato enzima di conversione dell'angiotensina (ACE). Nel 1982 Furakawa e collaboratori sintetizzarono i primi inibitori di ACE, inaugurando così un nuovo capitolo nella terapia dell'ipertensione. Nell'ultimo decennio i recettori AT1 dell'angiotensina II sono divenuti il bersaglio preferito di una nuova classe di farmaci antagonisti, i sartani, che attualmente rappresentano le armi di prima scelta nella terapia della malattia ipertensiva.
Nel 1975 John Hughes e Hans W. Kosterlitz isolarono dal cervello due pentapeptidi che riproducevano alcuni degli effetti della morfina, le encefaline, i primi oppioidi endogeni del cervello umano. Oggi si conoscono quattro famiglie di oppioidi endogeni: le encefaline, le endorfine, le endomorfine e le dinorfine. Sono stati identificati e clonati tre tipi di recettori oppioidi (μ, δ e κ) appartenenti alla famiglia dei recettori accoppiati alle proteine G, che comunicano l'informazione a effettori molecolari come l'adenilatociclasi e i canali del K+. La morfina, l'eroina e i principali oppiacei di sintesi agiscono tutti attraverso l'attivazione di questi recettori oppioidi. La scoperta del sistema oppioide endogeno ha apportato nuove conoscenze dei processi di dipendenza e di tolleranza rispetto ai farmaci oppiacei (tossicodipendenze). Nel 1985 Kelly A. Hickey e collaboratori descrissero un fattore vasocostrittore prodotto da cellule endoteliali in coltura, un peptide di ventuno amminoacidi denominato 'endotelina'. L'analisi genomica ha rivelato tre isoforme di endotelina, ET-1, ET-2 ed ET-3, e due principali sottotipi di recettori, ETA ed ETB. Sono stati sintetizzati alcuni farmaci antagonisti dei recettori dell'endotelina, che sono in corso di sperimentazione clinica nella terapia dell'ischemia renale e cerebrale.
Nell'ultimo ventennio del Novecento, lo sviluppo delle tecniche di sequenziamento del codice genetico ha permesso di definire le strutture primarie di grosse molecole peptidiche e di proteine coinvolte nelle numerose comunicazioni intra- e intercellulari. I fattori di crescita, le citochine, le chemochine, i fattori della coagulazione e dell'aggregazione piastrinica, la somatostatina, la calcitonina e numerose proteine intracellulari che mediano l'esecuzione dei messaggi ricevuti dai recettori sono esempi di macromolecole funzionali che rappresentano bersagli per nuovi farmaci o, direttamente, nuovi biofarmaci. Grazie alle biotecnologie di ingegneria genetica, alcune di queste proteine umane sono sintetizzate in larga scala dalla fabbrica cellulare, per essere utilizzate in terapia come tali o dopo avere introdotto opportune mutazioni nella loro sequenza peptidica per migliorarne la farmacodinamica o la farmacocinetica.
Recettori noti e recettori orfani: come nascono i nuovi farmaci
Considerati fin dall'inizio come il sito di azione dei farmaci, i recettori sono stati classificati per anni sulla base delle loro affinità e selettività farmacologiche; ma nell'ultimo ventennio del Novecento, le biotecnologie hanno permesso di identificare il codice genetico delle proteine recettoriali, ottenendo importanti informazioni sulle conformazioni possibili di queste macromolecole e su come esse siano trasportate e inserite nelle membrane cellulari. Con i progressi della strutturistica molecolare e l'ausilio degli elaboratori elettronici, i farmacologi hanno potuto disporre di modelli tridimensionali di recettori e di complessi farmaco-recettore. In alcuni casi le proteine recettoriali sono state isolate, purificate e perfino cristallizzate. Le tecniche fisiche di analisi (raggi X, risonanza magnetica nucleare e, per recettori giganti, la microscopia elettronica) hanno permesso di verificare direttamente la conformazione tridimensionale dei recettori isolati. Questo nuovo approccio di studio molecolare ha dimostrato che i recettori possono essere classificati in famiglie di proteine sufficientemente omogenee per organizzazione, topologia e dinamica molecolare, ma che presentano tra i loro membri un'ampia diversità molecolare. Sono stati così identificati molti nuovi tipi e sottotipi recettoriali per i quali i farmacologi non conoscevano, né, in molti casi, ancora conoscono, farmaci selettivi. In linea generale, i recettori dei farmaci possono trovarsi inseriti nella membrana cellulare o risiedere all'interno della cellula, nel citoplasma o nel nucleo. I principali recettori dei farmaci presenti sulla membrana citoplasmatica appartengono a distinte tipologie molecolari.
I canali ionici della membrana citoplasmatica
Il recettore del farmaco in questo caso è un canale ionico. L'apertura e la chiusura del canale possono essere regolate dal potenziale elettrico della membrana citoplasmatica (canali voltaggio-dipendenti), o direttamente dall'azione di un ligando endogeno su un sito recettoriale (recettori-canale). I tre principali canali voltaggio-dipendenti sono quelli dell'Na+, del K+ e del Ca2+. Il canale voltaggio-dipendente dell'Na+ è la sede di azione molecolare di farmaci antiepilettici, di anestetici locali e di antiaritmici. I canali ionici voltaggio-dipendenti per il Ca2+ sono componenti ubiquitari dei tessuti eccitabili di tutti gli organismi viventi. È possibile classificarli in canali a bassa soglia di apertura (si aprono per deboli depolarizzazioni di membrana) e canali a elevata soglia di apertura: i primi si inattivano rapidamente, i secondi rimangono aperti più a lungo. Le tre principali classi di farmaci antagonisti dei canali del Ca2+, le diidropiridine (nifedipina, nitrendipina, nicardipina, nimodipina), le benzodiazepine (Diltiazem) e le fenilalchilammine (Verapamil), si legano ai canali del calcio a elevata soglia di apertura. Questi farmaci, denominati dal loro meccanismo di azione molecolare 'calcioantagonisti', riducendo la corrente di ingresso di ioni calcio, funzionano come farmaci antiaritmici, antianginosi e ipotensivi.
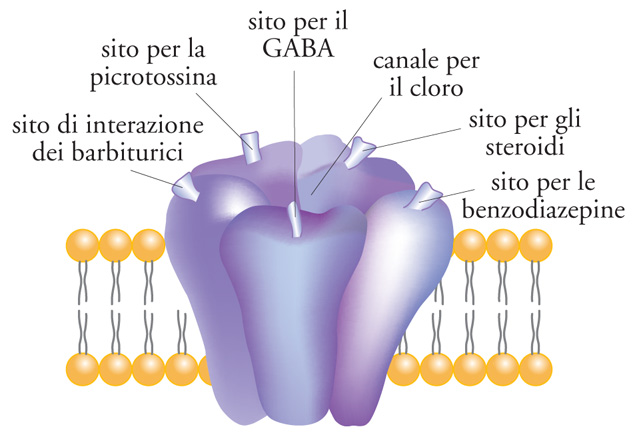
I canali del K+ rappresentano una classe molto eterogenea di canali ionici regolatori dell'eccitabilità cellulare. Se ne conoscono trenta tipi diversi, ma il loro numero è probabilmente destinato ad aumentare. La modulazione farmacologica di questi canali rappresenterà nel futuro il meccanismo molecolare più razionale per controllare l'eccitabilità cellulare. Alcuni farmaci antiaritmici, soprattuto quelli della classe III, si legano ai canali del K+ voltaggio-dipendenti del sistema di formazione e trasmissione del ritmo bioelettrico del cuore. Sulla membrana citoplasmatica sono localizzati alcuni canali ionici, la cui apertura è controllata da ligandi endogeni (recettori-canale). La selettività ionica di tutti questi recettori-canale non è molto spiccata per ioni con carica dello stesso segno, ma è assoluta per ioni di segno opposto. Di questi, le quattro famiglie più note sono i recettori-canale nicotinici, del glutammato, GABA-ergici e di tipo HT3. I recettori-canale nicotinici dell'acetilcolina conducono Na+ e K+ e svolgono nel sistema nervoso centrale un ruolo nei processi mnemonici. I recettori-canale del glutammato possono essere di due tipi: i recettori per l'AMPA e per l'acido cainico, responsabili della trasmissione rapida nelle sinapsi eccitatorie e che conducono prevalentemente Na+; i recettori per l'NMDA, coinvolti nella plasticità sinaptica e che conducono ioni Ca2+. Il GABA rappresenta il principale sistema di neurotrasmissione inibitoria; l'apertura dei canali di tipo A, che conducono Cl−, rende meno eccitabili i neuroni; agiscono su di esso l'etanolo, i barbiturici, le benzodiazepine (fig. 3). I recettori-canale di tipo 5-HT3 della 5-idrossitriptammina sono presenti in aree del sistema nervoso centrale implicate nel riflesso del vomito e conducono cationi (Na+, K+ e Ca2+).
A queste famiglie di recettori-canale modulati da ligandi endogeni se ne è recentemente aggiunta un'altra costituita da canali ionici che conducono K+ e che sono modulati dalle concentrazioni intracellulari di ATP. I canali di potassio modulati dall'ATP (K-ATP) hanno assunto recentemente una notevole importanza in quanto siti molecolari di interessanti azioni farmacologiche. Il legame con l'ATP ne determina la chiusura. Anche questa famiglia è assai eterogenea e comprende canali presenti nei miociti cardiaci, nella muscolatura liscia e scheletrica, in cellule nervose e nelle cellule β pancreatiche produttrici dell'insulina. Alcuni farmaci ipotensivi in uso da tempo nella terapia dell'ipertensione arteriosa, come il pinacidile, il minossidile e il diazossido, producono vasodilatazione periferica e caduta delle resistenze vascolari, aprendo i canali dei K-ATP delle fibre muscolari lisce vasali. I canali ionici di questo tipo sono normalmente chiusi o conducono poco, perché le concentrazioni di ATP intracellulari sono ben al di sopra di quelle necessarie per determinarne la chiusura. Ma in casi di anossia (per es., ischemia) le concentrazioni di ATP cadono sotto la soglia di chiusura e i canali dei K-ATP si aprono iperpolarizzando le membrane cellulari e producendo un arresto della contrazione e dell'attività bioelettrica: la cellula entra così in uno stato di risparmio energetico che, entro certi limiti, la protegge dal danno anossico. I farmaci capaci di aprire questi canali potenziano tale meccanismo fisiologico di protezione anossica e possono rappresentare una strategia terapeutica nell'infarto e nell'ictus cerebrale. Un'analoga azione a livello della muscolatura liscia bronchiale può essere utilizzata per prevenire attacchi di broncocostrizione in soggetti iperreattivi. Nelle cellule β pancreatiche i canali di K-ATP regolano la secrezione di insulina. I farmaci ipoglicemizzanti orali della classe delle sulfoniluree sono inibitori specifici dei canali K-ATP: attraverso tale meccanismo molecolare questi farmaci aumentano la secrezione di insulina in risposta al glucosio nei pazienti diabetici.
I recettori dell'insulina e dei fattori di crescita
Questo tipo di recettore è una proteina enzimatica di membrana che ha un dominio catalitico intracellulare (proteina-chinasi, guanilatociclasi) e un sito extracellulare di riconoscimento del ligando, connessi tra loro da una singola catena peptidica intramembranosa. A questa categoria appartengono i recettori dell'insulina e dei fattori di crescita e il recettore del peptide natriuretico atriale. Il ligando endogeno, o il farmaco, combinandosi con il sito extracellulare produce un cambiamento conformazionale che attiva il sito catalitico intracellulare o rende accessibile un sito del recettore che lega un enzima diffusibile, dando così inizio a una catena di reazioni enzimatiche di fosforilazione.
I recettori dei farmaci accoppiati alle proteine G
Il recettore di questo tipo è una macroproteina la cui catena di amminoacidi attraversa sette volte la membrana plasmatica. La porzione intracellulare del recettore è accoppiata a una proteina G. Le proteine G rappresentano una numerosa famiglia di proteine eterotrimeriche, di cui la subunità catalitica è capace di legare e idrolizzare il GTP. Questi recettori trasducono il messaggio chimico del ligando a molecole enzimatiche di membrana, quali la adenilatociclasi, la guanilatociclasi, la fosfolipasi, o a canali ionici. Un gran numero di farmaci e di trasmettitori endogeni si lega a recettori di questo tipo: appartengono a questa famiglia i recettori adrenergici di tipo α e β, i recettori dopamminergici, alcuni recettori della 5-idrossitriptammina, i recettori oppioidi, dell'angiotensina e di molti altri messaggi peptidici, i recettori di eicosanoidi e dell'istamina, i recettori metabotropici del glutammato, i recettori dei cannabinoidi.
Le pompe ioniche come siti molecolari di azione dei farmaci
Il recettore pompa ionica è un'unità catalitica di membrana che utilizza l'energia dei legami fosforici dell'ATP per pompare ioni fuori e dentro la cellula contro gradienti elettrochimici. Appartengono a questo tipo il recettore farmacologico dei glicosidi digitalici, la Na+/K+-ATPasi o 'pompa del sodio', e il recettore farmacologico dell'omeprazolo, la H+/K+-ATPasi o 'pompa protonica', presente nella membrana luminale dello stomaco, dove agisce come ultimo mediatore della secrezione acida. Na+ e ATP nella pompa del sodio e H+ e ATP nella pompa protonica si legano sulla faccia intracellulare delle pompe. I glicosidi digitalici e la forma attivata dell'omeprazolo, legandosi alle relative pompe, ne bloccano i cambiamenti conformazionali necessari per il funzionamento. I glicosidi digitalici agendo sulla pompa del sodio cardiaca esplicano la loro azione di aumento della forza di contrazione del cuore, che è utilizzata nella terapia dell'insufficienza cardiaca. L'omeprazolo, inibendo la pompa protonica gastrica, blocca la secrezione acida e trova pertanto impiego nella cura dell'ulcera gastroduodenale e nel reflusso esofageo.
I trasportatori di membrana come siti di azione molecolare dei farmaci
Il recettore in questo caso è un trasportatore di soluto che opera contro un gradiente e l'energia elettrochimica necessaria per il trasporto è fornita da un contro-ione. L'attenzione dei farmacologi è stata prevalentemente rivolta ad alcuni membri della famiglia dei trasportatori Na+/Cl−-dipendenti, che sono di importanza fondamentale per il processo di ricattura di neurotrasmettitori dopo la loro liberazione dai terminali presinaptici. Questa famiglia di trasportatori può essere suddivisa ulteriormente in due gruppi: i trasportatori delle monoammine (dopammina, 5-idrossitriptammina, noradrenalina), che sono il sito di azione molecolare di alcuni psicofarmaci come la cocaina, e i trasportatori degli amminoacidi (acido γ-amminobutirrico, glicina, betaina, taurina, prolina). Un'altra famiglia di trasportatori H+-dipendenti di interesse farmacologico è quella che concentra entro le vescicole sinaptiche i neurotrasmettitori monoamminici. Lo psicofarmaco reserpina si lega a questo sistema di trasporto, impedendo l'accumulo intravescicolare delle monoammine e privando i terminali sinaptici di questi neurotrasmettitori. In tal modo questi farmaci inibiscono la trasmissione sinaptica privando la sinapsi del neurotrasmettitore. Proteine trasportatrici di ioni sodio, potassio e cloro si trovano sulla membrana luminale delle cellule dei tubuli renali e funzionano da recettori di alcuni farmaci diuretici come la furosemide e i tiazidici.
I recettori nucleari
I recettori per gli ormoni steroidei (testosterone, estrogeni, progesterone, cortisonici) hanno invece sede intracellulare. Questi recettori, anche se hanno localizzazioni prevalenti diverse all'interno della cellula, posseggono tutti un elevato grado di omologia strutturale e di dinamica molecolare. I farmaci cortisonici liposolubili penetrano facilmente nella cellula e si legano al loro recettore disciolto nella parte fluida del citoplasma. In assenza dell'ormone, il recettore citoplasmatico forma un complesso molecolare inattivo con proteine da shock termico, come hsp90. In seguito all'interazione con l'ormone il recettore si distacca dalle proteine inibitorie, dimerizza e si trasferisce nel nucleo, dove, legandosi a specifiche sequenze di DNA presenti nei promotori di alcuni geni, ne modula la trascrizione. I farmaci antagonisti degli ormoni steroidei (antiandrogeni, antiestrogeni, spironolattoni) interagiscono con il recettore competendo con l'ormone naturale e operando il distacco del recettore dalle proteine inibitorie; il complesso recettore-antagonista però o non è in grado di legarsi alla sequenza del DNA (antagonismo di tipo I), oppure vi si lega ma non è poi capace di modificare la velocità di trascrizione del gene (antagonismo di tipo II).
Fino all'ultimo decennio del Novecento l'identificazione di tutti questi recettori era quasi sempre avvenuta utilizzando fondamentalmente due tecniche farmacologiche: lo studio delle risposte recettoriali ai farmaci su preparazioni isolate di tessuti e organi e, più recentemente, lo studio in vitro del legame di farmaci marcati con traccianti radioattivi a cellule o omogenati di cellule contenenti i recettori. Ma alla fine del Novecento e all'inizio del nuovo millennio la decodificazione del genoma ha rivelato una moltitudine inaspettata di nuove proteine recettoriali la cui funzione era, e in moltissimi casi è tuttora, ignota. Il DNA di questi recettori orfani è stato utilizzato per transfettare linee cellulari che, esprimendo, grazie al codice genetico ricevuto, selettivamente e in alta densità il recettore orfano, rappresentano un potente strumento per ricercare e individuare nuove molecole di agonisti e antagonisti. Questo approccio biotecnologico ha rivoluzionato la ricerca di nuovi farmaci offrendo un mezzo molto efficiente per saggiare l'attività di un gran numero di molecole in tempi brevissimi, senza l'uso di sperimentazione animale e in maniera completamente automatizzata. L'industria della strumentazione scientifica ha infatti immesso sul mercato macchine robotizzate che rivelano e quantificano l'attivazione dei recettori espressi da queste linee cellulari dopo l'aggiunta automatizzata di concentrazioni variabili di farmaci. Questi sistemi automatizzati di screening farmacologico, accoppiati al contemporaneo sviluppo della sintesi chimica in fase solida e della chimica combinatoriale, hanno permesso di analizzare librerie chimiche di migliaia di prototipi di potenziali farmaci e di rivelare le funzioni di molti recettori orfani. La farmacologia molecolare si è così progressivamente trasformata da scienza di studio del recettore in strumento di identificazione di nuovi farmaci.
Geni e farmaci
Alla fine del Novecento si è consolidata un'altra fondamentale acquisizione sul meccanismo d'azione dei farmaci: l'azione di molti farmaci non si esaurisce nell'immediata risposta recettoriale che determina l'effetto farmacologico, ma si estende nel tempo attraverso una modulazione della trascrizione genica. La farmacologia molecolare ha fatto progredire le nostre conoscenze nell'ambito della superfamiglia dei recettori accoppiati a fattori di trascrizione genica, come per esempio ai fattori di trascrizione della famiglia AP-1 (proteine Fos e Jun), la cui trascrizione viene indirettamente attivata da segnali provenienti dalla membrana citoplasmatica a seguito della reazione farmaco-recettore. Con processi di indiretta modulazione dell'espressione genica, attraverso un complesso sistema di trasduzione intracellulare del segnale recettoriale evocato dal farmaco, la somministrazione ripetuta di un farmaco può determinare modifiche del fenotipo. Ciò ha evidenti implicazioni sia terapeutiche che tossicologiche e sembra rivestire particolare importanza nella terapia con psicofarmaci e nei processi di tossicodipendenza. Ma gli stessi fattori di trascrizione possono essere bersaglio di farmaci. Un esempio è offerto da un vecchio farmaco, l'aspirina. Mentre l'aspirina, somministrata a piccole dosi ‒ sufficienti per rimuovere dolori lievi, ridurre la febbre e inibire l'aggregazione piastrinica ‒ svolge il suo effetto terapeutico attraverso l'inibizione della ciclossigenasi e il conseguente blocco della sintesi dei prostanoidi infiammatori, a dosi più elevate, quali quelle utilizzate nella terapia dell'artrite reumatoide, agisce a livello di un fattore di trascrizione genica, il fattore NFκB, che esplica la sua azione regolando la trascrizione di proteine importanti nella patogenesi della malattia infiammatoria.
La terapia genica del futuro ripone molte speranze di successo nello sviluppo di molecole capaci di attivare un gene, di reprimerlo o di trasferire nelle cellule malate del paziente un gene modificato. In teoria ciò è possibile con due diversi tipi di tecniche: terapia genica ex vivo e terapia genica in vivo. Nella terapia genica ex vivo le cellule del paziente in cui si deve spegnere l'espressione di un gene o in cui si vuole attivare la trascrizione di un gene vengono isolate e coltivate in vitro. Il gene bersaglio viene bloccato o attivato transfettando le cellule in coltura con opportuno materiale genico e le cellule transfettate vengono reinserite nello stesso paziente. Il vantaggio di questa tecnica è che esiste già una notevole esperienza nella transfezione genica in colture cellulari. Le difficoltà risiedono nelle tecniche di impianto e nel funzionamento di queste cellule modificate una volta reinserite nel paziente, e nel costo dell'intero procedimento che è comunque strettamente individuale. Questa tecnica ha già avuto riscontri positivi in alcune leucemie, nella malattia di Parkinson e nell'infarto del miocardio. Nella terapia genica in vivo il gene terapeutico, o il soppressore del gene malato, viene inserito nelle cellule bersaglio iniettandolo direttamente nel paziente. Questa tecnica è ai primi stadi sperimentali negli animali di laboratorio. Anche se la farmacologia di oggi è ancora in larga parte la farmacologia degli agonisti e degli antagonisti dei recettori biologici, quella di domani sarà rivolta a modulare le funzioni biologiche del paziente, inibendo o esaltando l'espressione genica delle molecole endogene responsabili della funzione.
La presenza di variazioni nella sequenza del DNA di uno stesso gene nei diversi soggetti umani è un fenomeno ben noto e quando una tale variazione è presente in più dell'1% della popolazione si parla di polimorfismo genico. I geni che determinano la risposta terapeutica ai farmaci possono essere distinti in due grandi classi: i geni codificanti per le proteine che mediano l'azione del farmaco (recettori) e i geni codificanti per le proteine che regolano la farmacocinetica (assorbimento, trasporto, metabolismo, eliminazione del farmaco). Il polimorfismo dei geni regolatori della risposta terapeutica è la causa prima della variabilità dei risultati terapeutici ottenuti con i farmaci nelle diverse malattie. Le tecniche di analisi genomica permettono di identificare e localizzare nei geni le variazioni responsabili della variabilità terapeutica. Ne sono esempi l'elevato numero di variazioni riscontrate nei geni codificanti per la superfamiglia di proteine del citocromo P450, che metabolizzano nel fegato un gran numero di farmaci, le varianti alleliche riscontrate nell'enzima umano N-acetiltransferasi, che acetila molti farmaci, e le variazioni nel codice genetico della tiopurina-metiltransferasi, una proteina enzimatica che inattiva le tiopurine, farmaci adoperati nella terapia antineoplastica. È evidente che identificando le variazioni nel genoma del singolo paziente si può cambiare il farmaco interessato dalla variazione e utilizzato per la cura con uno che non sia interessato dal polimorfismo genico. Si prospetta così nel futuro una terapia personalizzata che, attraverso l'analisi del genoma del singolo paziente e la produzione di farmaci modificati, eviti opportunamente gli inconvenienti terapeutici prodotti dal polimorfismo genico.
Bibliografia
Clementi, Fumagalli 2004: Clementi, Francesco - Fumagalli, Guido, Farmacologia generale e molecolare, 3. ed., Torino, UTET, 2004.
Howard 2001: Howard, Andrew D., Orphan G-protein-coup_led receptors and natural ligand discovery, "Trends in pharmacological sciences", 22, 2001, pp. 130-140.
Kenakin 2004: Kenakin, Terry P., Principles: receptor theory in pharmacology, "Trends in pharmacological sciences", 25, 2004, pp. 186-192.
Tinsley, Eriksson 2004: Tinsley, Rogan - Eriksson, Peter, Use of gene therapy in central nervous system repair, "Acta neurologica scandinavica", 109, 2004, pp. 1-8.