distrofie muscolari
distrofie muscolari
Miopatie progressive geneticamente determinate, in cui il tessuto muscolare, per effetto dei processi di necrosi e di rigenerazione delle fibre, presenta fibrocellule ipertrofiche con nuclei centrali, aumento del tessuto fibroso connettivo e sostituzione adiposa del tessuto muscolare. Sono in gran parte dovute a mutazioni di geni che codificano proteine del sarcolemma (distrofina, sarcoglicani, disferlina) e della membrana nucleare (emerina, lamina A/C), enzimi (calpaina), proteine sarcomeriche (miotilina) e della matrice extracellulare (laminina).
Distrofinopatie
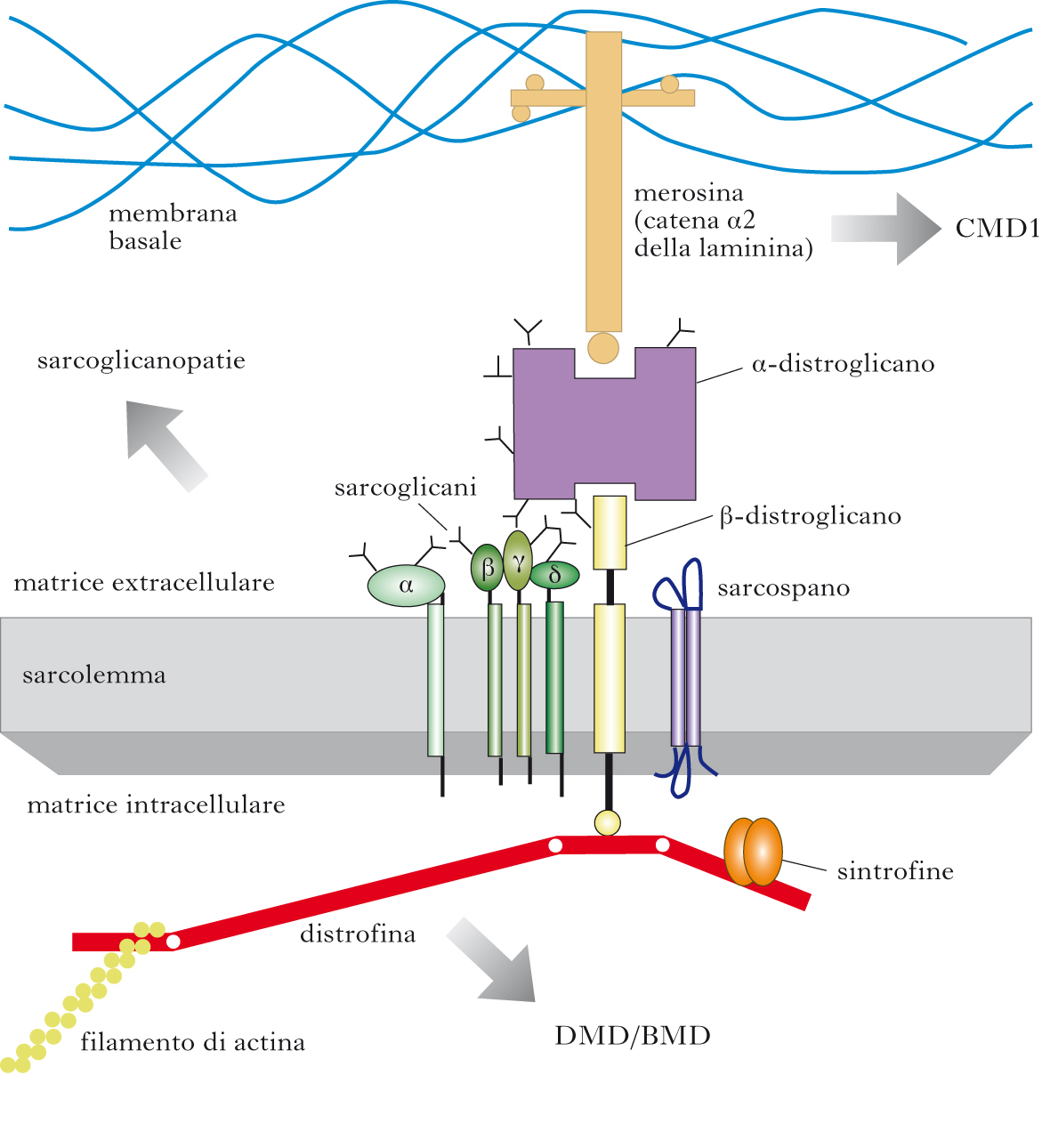
Sono d. m. recessive, da deficit di distrofina, proteina codificata dal gene Xp21 ed espressa nel muscolo scheletrico, cardiaco e liscio, nella corteccia cerebrale, nell’ippocampo e nelle cellule del Purkinje. Localizzata tra sarcoplasma e sarcolemma, la distrofina, la cui parte terminale lega i filamenti di actina, è ancorata al sarcolemma tramite complessi glicoproteici DAPs (Dystrophin-Associated Proteins), comprendenti distroglicani (α e β), sarcoglicani (α, β, γ e δ) e sintrofine. Il deficit di distrofina altera la stabilità meccanica del sarcolemma durante la contrazione-rilasciamento; ciò favorisce un danno della membrana, con massiccia entrata di calcio nella cellula, attivazione di enzimi proteolitici e necrosi della cellula. Si distinguono due forme cliniche: la d. m. di Duchenne (➔) (DMD) a esordio più precoce e più grave, e la d. m. di Becker (➔) (BMD) a esordio più tardivo e meno grave. Mentre nella DMD la distrofina è quasi completamente o del tutto assente, nella BMD è di alterato peso molecolare e quantitativamente ridotta (85% dei casi) o di peso molecolare normale, ma quantitativamente ridotta (15% dei casi). La quantificazione della distrofina con immunoblot è predittiva della gravità della malattia: riduzione superiore al 95% nella DMD, riduzione fra il 95% e l’80% in forme intermedie fra DMD e BMD, riduzione inferiore all’80% nella BMD lieve/moderata. La mutazione genetica può consistere in una delezione (60÷65%), una duplicazione (5%) o una mutazione puntiforme (2/3 del rimanente 30%). L’espressione della distrofina è influenzata non dall’entità della delezione, ma dalla conservazione o dalla distruzione del quadro di lettura per effetto della delezione stessa, le quali generano rispettivamente una distrofina semifunzionale (BMD) o una distrofina gravemente troncata (DMD). Il 30% dei casi di distrofinopatia (DMD, in partic.) è dovuto a nuova mutazione. Occasionalmente le femmine portatrici della mutazione genetica possono essere affette dalla malattia. Ciò accade nella sindrome di Turner, oppure nel caso in cui, durante il processo di lyonizzazione, avvenga un’inattivazione sbilanciata, nella gran parte delle cellule embrionali, di uno dei due cromosomi X a favore di quello paterno, per cui meno del 50% delle cellule mature esprime distrofina funzionale.
Distrofia muscolare di Emery-Dreifuss (EDMD)
Presenta una triade sintomatologica: contratture muscolari precoci (muscoli estensori del collo e della colonna con spina rigida, del bicipite e della sura), debolezza e atrofia scapolo-omero-peroneale, e cardiomiopatia. È una d. legata al cromosoma X, dovuta a deficit di emerina o lamina A/C.
Distrofie muscolari congenite (CMD)
Hanno esordio precoce (congenito o entro un anno) con debolezza diffusa (floppy baby), contratture e deformità scheletriche (cifoscoliosi), disturbi a carico del sistema nervoso centrale dovuti a difetti di mielinizzazione o a migrazione neuronale. Hanno decorso variabile, da forme a esito infausto nei primi anni di vita, fino a forme stabilizzate nell’adulto. Esistono cinque tipi clinici di CMD: la CMD1, più comune in Europa, da deficit di merosina, presenta marcato incremento dell’enzima creatininchinasi, grave ipotonia neonatale, ritardo dello sviluppo motorio, contratture muscolari e insufficienza respiratoria, marcate alterazioni della sostanza bianca, normale sviluppo mentale; la CMD2 (detta anche malattia di Fukujama), più comune in Giappone, è una sarcoglicanopatia da deficit di fukutina, che interviene nella glicosilazione dell’α-sarcoglicano e nella migrazione neuronale; la CMD3, da difetto del collagene 6 e delle selenoproteine N, ha caratteristiche analoghe alla CMD1 (ma con gravi deficit psicointellettivi), dà luogo a gravi contratture e debolezza dei muscoli prossimali e distali, e si manifesta in due forme cliniche, una più grave (miopatia di Ullrich) e l’altra più lieve (miopatia di Bethlem); la CMD4, da deficit di integrina alfa7.
Distrofie muscolari dei cingoli (LGMD)
Forme di d. che danno debolezza lentamente progressiva e simmetrica dei muscoli prossimali degli arti, integrità dei muscoli facciali, e possibile associazione di cardiomiopatie e di deficit respiratori. Più frequenti le forme a trasmissione autosomica recessiva (LGMD2) rispetto a quelle dominanti (LGMD1). Le LGMD2 sono dovute ad alterazioni di proteine di membrana (sarcoglicani, disferlina), della matrice extracellulare (laminina), proteine strutturali e contrattili (titina) e del sarcolemma (fukutina). Le LGMD1 sono dovute ad alterazione di proteine connesse con il sarcolemma (caveolina), con la membrana nucleare (lamina a/c) e con il sistema contrattile (miotilina).

Distrofia facio-scapolo-omerale (FSHD)
È una d. m. autosomica dominante, provocata da una delezione della sequenza D4Z4 sul cromosoma 4. È caratterizzata da debolezza dei muscoli del volto e del cingolo scapolare, frequente coinvolgimento dei muscoli peroneali, pettorali e addominali (segno di Beevor) spesso asimmetrico con risparmio dei muscoli deltoidi, faringo-laringei, extraoculari e respiratori. Questa forma di distrofia è spesso accompagnata da ipoacusia neurosensoriale (75% dei casi) e da vasculopatie retiniche (60%). Si ha perdita dell’autonomia motoria in un quinto dei casi.
Distrofia miotonica di Steinert
D. m. a trasmissione autosomica dominante, caratterizzata da miotonia associata a debolezza muscolare (➔ Steinert, malattia di).
Distrofia oculo-faringea
Rara d. m. generalmente a trasmissione autosomica dominante, per un’espansione GCG nel gene per la PAB2 (Poly A Binding protein-2) sul locus 14q11. È caratterizzata da debolezza dei muscoli faringolaringei e da oftalmoplegia.