
Duchenne, distrofia di
Duchenne, distrofia di
Malattia genetica, descritta clinicamente e istopatologicamente dal neurologo francese Guillaume-Benjamin Duchenne, caratterizzata da una trasmissione ereditaria, legata al cromosoma X, di un gene recessivo, che codifica una proteina chiamata distrofina. L’assenza della distrofina causa degenerazione del tessuto muscolare, progressiva perdita di forza muscolare e riduzione delle abilità motorie. È una distrofia (➔) uniformemente diffusa in Europa, Stati Uniti, Giappone e Australia, con un‘incidenza di 15÷30 casi su 100.000 nati maschi e una prevalenza di 30÷40 individui su ogni milione di abitanti. Il gene della distrofia di D. è localizzato nel braccio corto del cromosoma Xp21; la distrofia muscolare origina talora da una microdelezione, in altri casi vi è la presenza di una mutazione che altera o modifica il codice di lettura dell’RNA messaggero. Nella maggior parte dei casi il gene viene trasmesso ai figli maschi affetti dalla madre portatrice della malattia. Le portatrici hanno nel 70% dei casi alti livelli dell’enzima creatinfosfochinasi (CPK), e nella biopsia muscolare una distribuzione a mosaico, con fibre positive per la distrofina alternate a fibre negative.
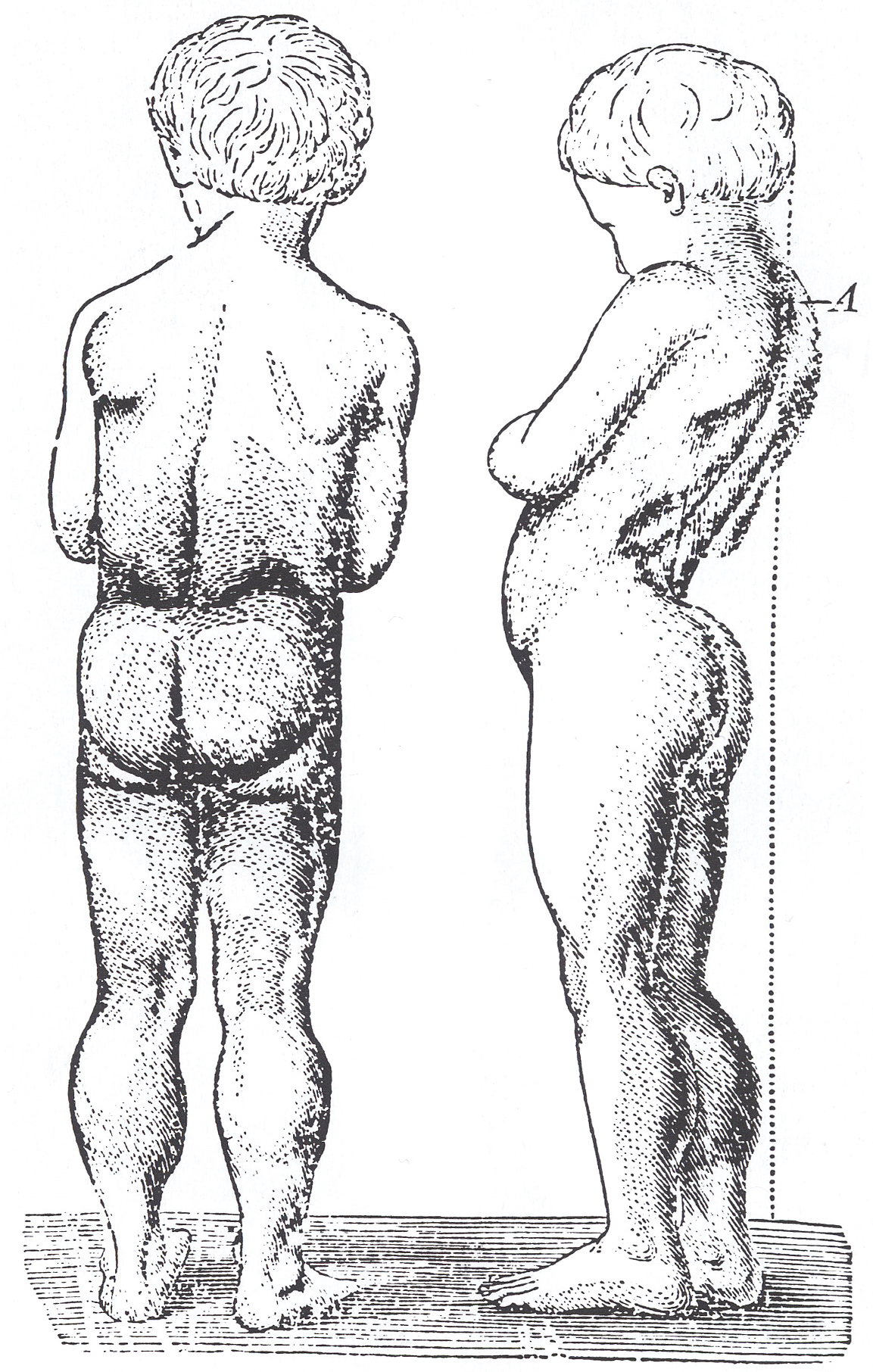
Sintomi
Il paziente con la distrofia di D. presenta facilità alla caduta a 2÷3 anni, una deambulazione anserina (come un’anatra) e, nel rialzarsi da terra, mostra la manovra di Gowers, ossia appoggia le mani al pavimento o sulle ginocchia; dopo i 10 anni è spesso incapace di deambulare autonomamente. L’intelligenza è compromessa in alcuni casi, benché tale alterazione non sia progressiva. Nei pazienti obbligati alla carrozzina, il mantenimento della stazione seduta provoca scoliosi, insufficienza respiratoria e contratture con anchilosi delle ginocchia e caviglie. Nella seconda decade di vita il paziente va incontro a cardiopatia (spec. cardiomiopatia dilatativa), obesità, cifoscoliosi, retrazioni tendinee e complicazioni polmonari, spesso causa del decesso.
Diagnosi
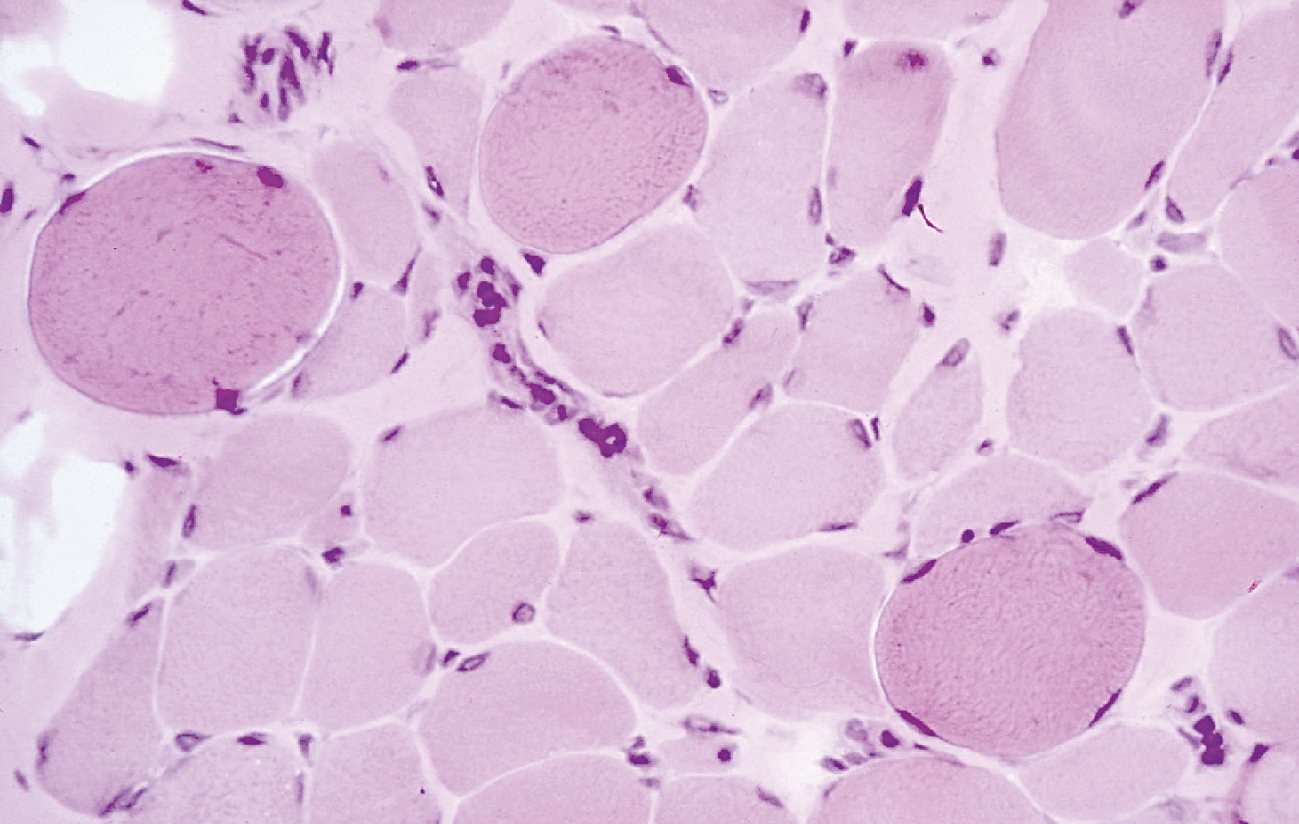
La diagnosi differenziale si effettua tramite biopsia muscolare ed elettromiografia, anche se le tecniche molecolari permettono una diagnostica sul DNA nel 60% dei casi. Alla biopsia muscolare gli aspetti morfologici del muscolo scheletrico sono caratterizzati dalla comparsa di necrosi a piccoli gruppi di fibre muscolari; nelle fibre in necrosi, la reazione tricromica (metodo elettivo di colorazione istologica per il connettivo e per le fibre del connettivo) può evidenziare un aspetto più denso del sarcoplasma oppure, se vi è deposito di calcio, fibre contratte chiamate fibre opache. Nelle fasi avanzate alla sclerosi del tessuto connettivo si associa la sostituzione con tessuto adiposo. La conferma diagnostica dei reperti della biopsia muscolare è data dall’assenza della distrofina, valutata sia con l’immunoistochimica sia con il western blot (tecnica immunochimica che permette di identificare la presenza di una determinata proteina in una serie di proteine separate elettroforeticamente, mediante il riconoscimento da parte di anticorpi specifici). In alcune fibre (dette fibre revertite) è possibile che spontaneamente si arrivi a una retromutazione somatica che permette la risintesi della distrofina. Con l’adozione di adatte tecniche è possibile la diagnosi di eterozigosi nelle donne portatrici e la diagnosi prenatale in donne che non hanno casi affetti in famiglia: si adottano sia il metodo con PCR multiplex sia, nella biopsia muscolare, l’espressione a mosaico della distrofina.
Terapia
L’unico trattamento farmacologico palliativo è costituito dagli steroidi, terapia studiata con numerose prove terapeutiche randomizzate e con diversi protocolli di somministrazione. In vari Paesi sono in fase di avanzata sperimentazione umana gli oligonucleotidi antisenso che permettono di bypassare l’esone alterato o terapie sostitutive con cellule staminali. Queste nuove possibilità terapeutiche probabilmente richiederanno molti anni di sperimentazione. Un grande impegno va diretto quindi a una terapia di sostegno riabilitativa i cui cardini sono:
• Prevenzione dell’aumento di peso e dell’obesità del paziente.
• Profilassi della cardiopatia, che consiste nell’uso di ACE-inibitori, diuretici, beta-bloccanti in fasi precoci. Se esistono turbe aritmiche importanti, si installano pacemaker, con o senza funzione di defibrillatore.
• Modica attività muscolare e fisioterapia respiratoria; la riabilitazione fisica e la massoterapia sono utili nel ritardare le contratture muscolari nelle fasi tardive.
• Aiuto ventilatorio; aiuto psicologico ai bambini e ai genitori; per le complicanze respiratorie si ricorre precocemente alla ventilazione per via nasale notturna con mascherina a pressione positiva; questi presidi permettono un prolungamento della vita del paziente, che può giungere così alla 3ª÷4ª decade di vita. È importante fornire al paziente distrofico e ai familiari consigli riguardo al suo inserimento scolastico e provvedere alla sua autonomia di spostamento con ortesi e carrozzine. Allo scopo di migliorare l’inserimento sociale dei pazienti distrofici è sorta in Italia la UILDM (Unione Italiana Lotta Distrofia Muscolare) che si adopera con la Fondazione Telethon per la ricerca in questo campo.