
Dinamica molecolare
Dinamica molecolare
La ricerca nel campo della dinamica molecolare ha come obiettivo lo studio del livello microscopico dei processi di trasformazione delle sostanze. Essa si propone di chiarire in che modo le grandezze macroscopiche osservabili, che descrivono tali processi, dipendano dalla distribuzione, nei reagenti e nei prodotti delle reazioni relative, di alcune grandezze fisiche microscopiche, come la velocità, il momento della quantità di moto e l'energia associata ai gradi di libertà interni (rotazionali, vibrazionali, elettronici) delle molecole reagenti e di quelle prodotte. Per quanto concerne la reattività, le grandezze osservabili, quali le costanti di velocità e le sezioni d'urto, possono essere misurate con un grado di accuratezza che dipende dal sistema in esame e dall'avanzamento delle tecniche sperimentali.
Negli ultimi decenni, il progresso in questo campo ha consentito di elaborare teorie e tecniche computazionali che permettono di correlare i fenomeni sensibili con le forze d'interazione intermolecolari (superfici di energia potenziale), verificando ed estendendo i semplici modelli della cinetica chimica. Per illustrare alcuni aspetti dell'ampia fenomenologia ormai nota, considereremo alcuni esempi di processi elementari in fase gassosa, sufficientemente rappresentativi sia delle tecniche sperimentali e interpretative in uso, sia delle prospettive e dei problemi relativi alla modellizzazione di processi macroscopici a partire dalla dinamica dei processi elementari microscopici. Lo studio della velocità delle reazioni chimiche e della dinamica molecolare risulta di primaria importanza non solo dal punto di vista della ricerca fondamentale ‒ per approfondire la conoscenza della struttura della materia e delle leggi che la governano ‒ ma anche da quello applicativo; in questo secondo caso, esso apre prospettive importanti per la preparazione di nuovi composti e per la comprensione dell'evoluzione di sistemi complessi (processi di combustione, chimica e fisica delle atmosfere planetarie e dei plasmi, comportamento della materia in condizioni estreme di pressione e di temperatura).
La cinetica delle reazioni è diventata un capitolo importante della chimica a partire dalla metà dell'Ottocento. All'inizio, l'attenzione è stata rivolta soprattutto alla determinazione delle leggi che governano le velocità delle reazioni e la loro dipendenza da grandezze quali le concentrazioni delle specie reagenti e la temperatura del sistema. Successivamente, le ricerche hanno riguardato la determinazione del meccanismo delle reazioni chimiche, vale a dire la caratterizzazione di reazioni complesse come combinazione di processi elementari. Lo stadio più avanzato, infine, è consistito nell'interpretazione del processo macroscopico secondo un modello dettagliato di procedimenti microscopici. Fin dalla metà dell'Ottocento, parallelamente alla nascita della teoria cinetica dei gas, è risultato evidente il ruolo delle collisioni tra molecole reagenti nelle reazioni bimolecolari; di contro, si è dovuto attendere il primo quarto del XX sec. per interpretare le reazioni monomolecolari in termini di trasferimento intermolecolare e di ridistribuzione dell'energia nei moti intramolecolari.
La legge generale che descrive la dipendenza dalla temperatura termodinamica della costante o coefficiente di velocità ‒ formulata dallo svedese Svante A. Arrhenius nel 1889 ‒ prevede una dipendenza esponenziale da un parametro empirico, l'energia di attivazione, definita come la quantità minima di energia di collisione che le molecole devono possedere per poter reagire. L'equazione di Arrhenius k=Ae−E/RT (dove k è la costante o coefficiente di velocità, E l'energia di attivazione, A il fattore di frequenza, R la costante universale dei gas, T la temperatura termodinamica), può essere quindi considerata l'atto di nascita dello studio della dinamica delle reazioni chimiche, intesa come studio del moto degli atomi e delle molecole nel singolo evento reattivo.
Nella prima metà del secolo scorso la dinamica delle reazioni chimiche ha acquisito solide basi teoriche, grazie alla meccanica quantistica. A partire dagli anni Sessanta si sono rese disponibili le tecniche sperimentali che permettono lo studio dettagliato delle reazioni a livello microscopico. Da allora, la dinamica molecolare è diventata uno dei settori di punta della scienza chimica. La sua importanza è stata riconosciuta con l'assegnazione del premio Nobel per la chimica nel 1986 a Dudley R. Herschbach, Yuan T. Lee e John C. Polanyi, e nel 1999 ad Ahmed H. Zewail.
Aspetti sperimentali
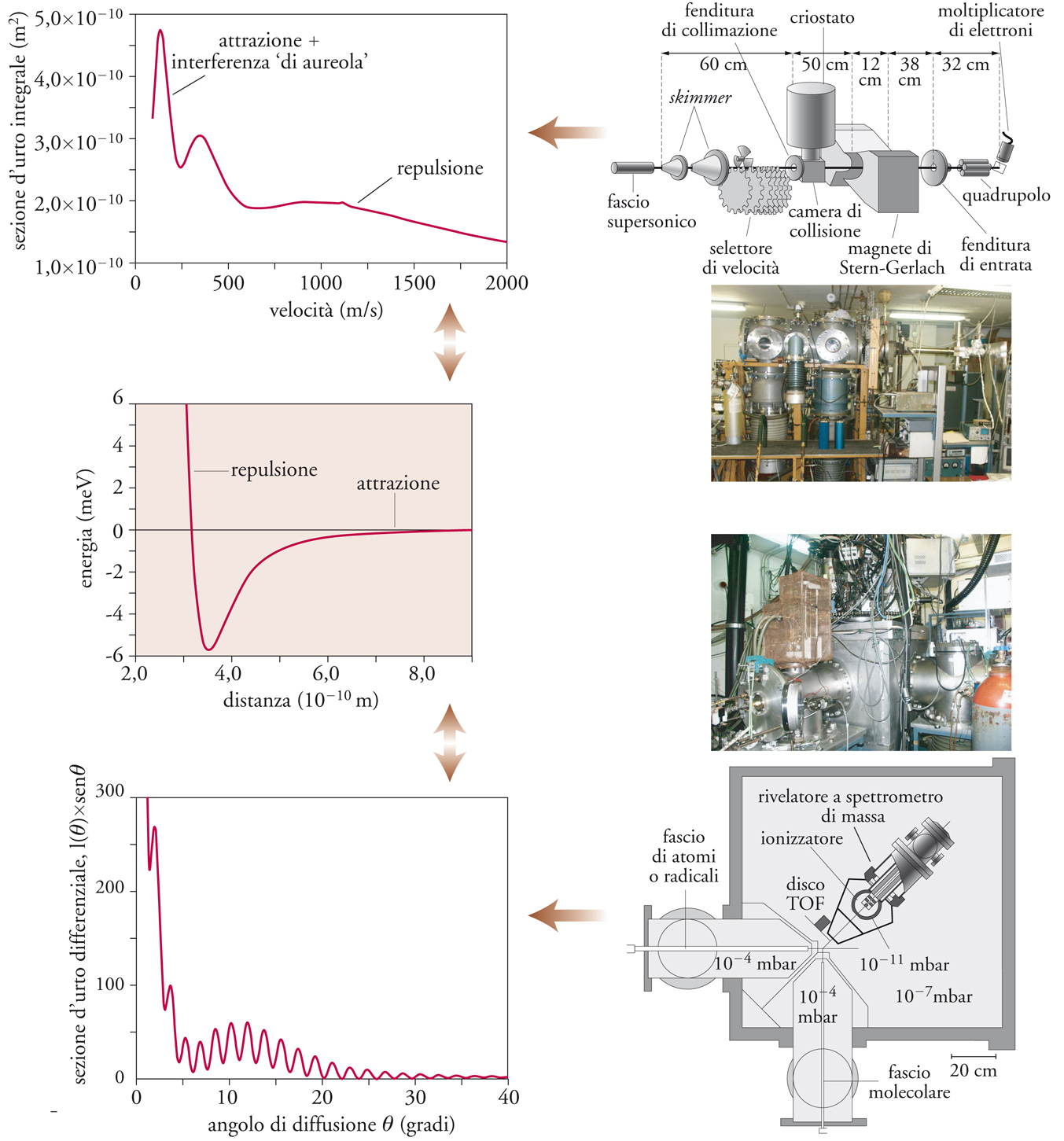
Le tecniche sperimentali utilizzate per lo studio della dinamica delle reazioni chimiche possono essere classificate in due grandi categorie: collisionali e spettroscopiche. Le prime (Tav. I) consentono di ottenere valori sperimentali delle sezioni d'urto integrali o differenziali, che forniscono una misura della probabilità che una collisione tra particelle si risolva in un evento reattivo. Uno degli aspetti più interessanti di tali esperimenti consiste nel fatto che essi permettono di verificare, mediante un confronto più o meno diretto con il dato sperimentale, l'accuratezza dei modelli teorici e dei metodi computazionali. Una caratteristica importante delle tecniche collisionali è, in particolare, quella di permettere la misura della distribuzione angolare dei prodotti di reazione. L'apparecchiatura per misure differenziali mostrata nella Tav.I (in basso a destra) consente anche lo studio di reazioni, per le quali si possono ottenere informazioni sulla ripartizione dell'energia tra moto traslazionale e moti interni attraverso la determinazione, con una tecnica detta a tempo di volo o TOF (Time of flight), della velocità dei prodotti. Questa tecnica venne usata per la prima volta con successo nello studio di una reazione chimica da Ellison H. Taylor e Sheldon Datz nel 1955 negli USA, ed è stata sviluppata e utilizzata per lo studio di una notevole varietà di reazioni dal gruppo di ricerca di Herschbach negli anni Sessanta e Settanta e da quello di Lee negli anni Settanta e seguenti. Le tecniche di produzione dei fasci variano a seconda della natura delle specie in gioco. In genere, uno dei reagenti è una specie chimica stabile, cosicché il fascio molecolare può essere prodotto per semplice espansione del gas attraverso un foro oppure una fenditura in una camera da vuoto, con successiva collimazione per mezzo di altre fenditure. In tal modo si ottiene un fascio di particelle che procedono in una determinata direzione senza subire collisioni con il gas residuo.
In un esperimento di dinamica di una reazione è praticamente impossibile controllare contemporaneamente i numerosi parametri che entrano in gioco, quali la velocità, gli stati interni e l'orientazione dei reagenti e dei prodotti. Sono quindi necessari processi di media, ciascuno dei quali toglie parte delle informazioni contenute nell'esperimento: per esempio, nelle sezioni d'urto differenziali, si considera una media tra specifici stati rotazionali, vibrazionali ed elettronici dei reagenti e dei prodotti. Si ottengono, in questo modo, sezioni d'urto differenziali o integrali per insiemi di particelle, per i quali alcuni dei gradi di libertà possono non essere univocamente definiti. Sebbene ciò complichi l'interpretazione degli esperimenti, è innegabile che le misure di sezioni d'urto contengono sempre qualche informazione supplementare rispetto alle misure tradizionali delle costanti di velocità delle reazioni. Queste ultime implicano la possibilità di definire la temperatura e quindi l'equilibrio termico di tutti i gradi di libertà traslazionali e interni dei reagenti e dei prodotti. Pertanto, le costanti di velocità sono sempre ottenibili, almeno in via di principio, mediando le sezioni d'urto secondo le distribuzioni di Boltmann su quei gradi di libertà che non si è riusciti a controllare. Il processo inverso (e cioè una deconvoluzione da costanti di velocità a sezioni d'urto) non è invece praticabile. Anzi, è necessario introdurre esplicitamente le sezioni d'urto per interpretare le velocità dei processi reattivi o di trasporto nei casi in cui l'ipotesi dell'equilibrio termico non sia verificata, vale a dire nelle condizioni in cui la temperatura non sia un parametro significativo.
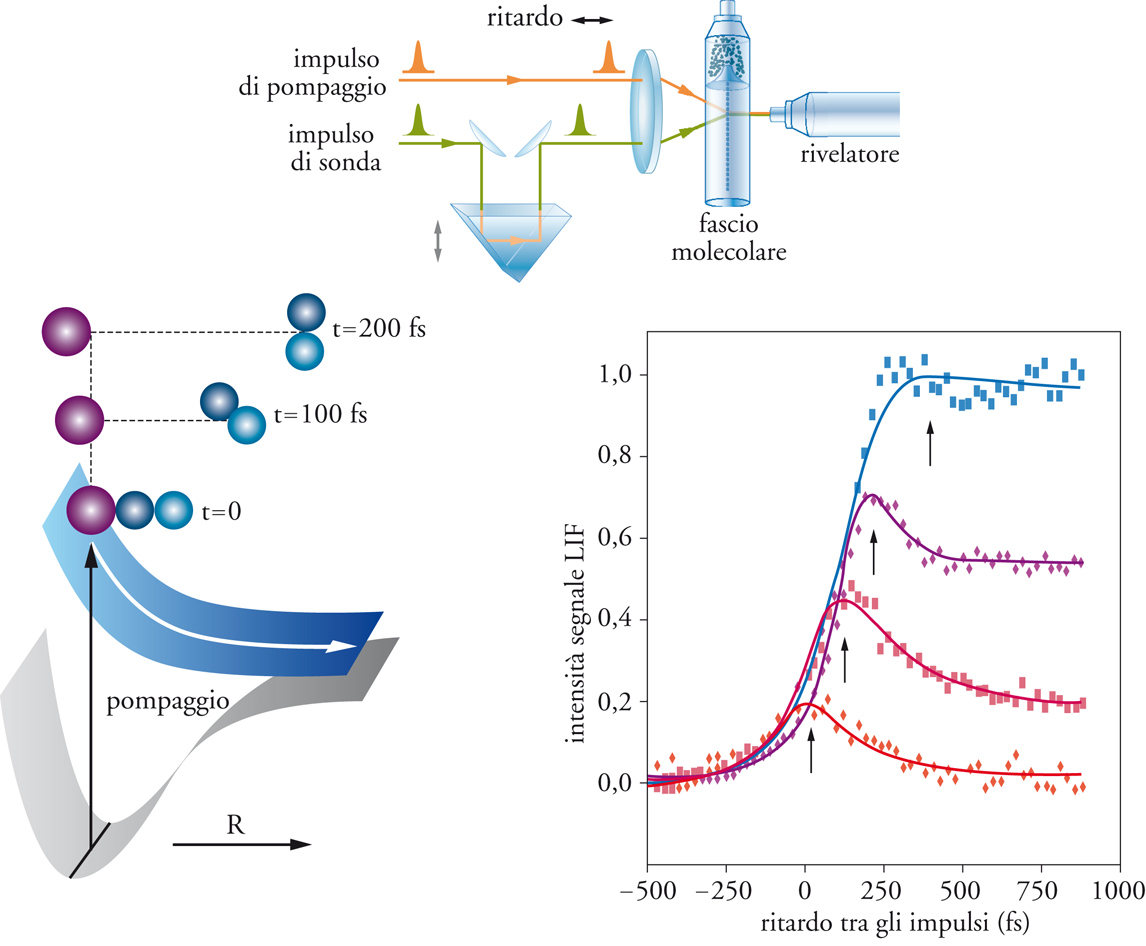
La seconda grande famiglia di metodiche sperimentali utilizzate per lo studio della dinamica delle reazioni chimiche è rappresentata dalle tecniche spettroscopiche, che permettono di seguire l'evoluzione nel tempo dei reagenti nei prodotti e di determinare lo stato energetico interno delle specie coinvolte. Un esempio tipico è rappresentato dalla misurazione della chemiluminescenza nell'infrarosso dei prodotti nascenti, cioè di prodotti che si trovano a pressioni tanto basse e che vengono rivelati a tempi così brevi dalla loro produzione, da non poter subire collisioni prima di emettere la radiazione caratteristica dei loro stati interni. Mediante questa tecnica, applicata fin dal 1958 da Michael Polanyi, è possibile ricavare la popolazione degli stati eccitati vibrorotazionali risultanti da particolari processi reattivi. Più generale è l'utilizzazione della fluorescenza indotta da laser o LIF (Laser induced fluorescence), che viene applicata a sistemi caratterizzati da stati eccitati con vita media molto breve. In seguito, sono state sviluppate altre tecniche basate sull'estensione dei metodi spettroscopici (Tav. II). Oltre a queste tipologie limite di tecniche spettroscopiche, esiste una serie di tecniche sperimentali intermedie, che spesso possono essere utilizzate per lo studio di reazioni specifiche. Per esempio, con le tecniche di spettroscopia veloce, impiegando impulsi di pompaggio e di sonda linearmente polarizzati e analizzando lo spostamento Doppler della fluorescenza indotta dal laser della pompa, si possono ottenere informazioni sulla direzione del moto dei prodotti. Le reazioni nelle quali sono coinvolte specie cariche hanno caratteristiche particolari. Gli ioni reagenti o quelli prodotti possono essere selezionati ‒ più facilmente che non le specie neutre ‒ mediante spettrometria di massa, e l'energia di collisione può essere variata in intervalli piuttosto ampi, soprattutto per energie superiori all'energia termica. Infine, l'impiego di laser polarizzati negli impulsi di pompaggio e di sonda apre notevoli prospettive per lo studio della stereospecificità delle reazioni in funzione dell'orientamento dei reagenti o per l'analisi dell'orientamento dei prodotti.
Aspetti teorici
Le proprietà di trasporto della materia (quali la conducibilità e la viscosità) e le reazioni chimiche sono manifestazioni macroscopiche di collisioni che avvengono a livello atomico e molecolare, e sono descritte, in ultima analisi, dalle leggi generali che regolano le collisioni. Tali leggi sono quelle della meccanica quantistica e, per lunghezze d'onda molto piccole, della meccanica classica. Tuttavia, data la complessità dei sistemi di interesse pratico, l'applicazione di queste leggi è ostacolata da enormi difficoltà di calcolo, dovute principalmente al fatto che le equazioni del moto dipendono da un numero elevatissimo di coordinate (tre per ogni elettrone e tre per ogni nucleo). D'altra parte, fino alla metà del XX sec., le informazioni sperimentali disponibili erano fornite da esperimenti su proprietà macroscopiche, e quindi costituite da quantità, come i coefficienti di trasporto e le costanti di velocità, mediate su un numero enorme di collisioni, le cui proprietà medie possono, in casi favorevoli, corrispondere a situazioni di equilibrio caratterizzate da una temperatura ben definita. Pertanto, le approssimazioni dei modelli usati in quegli anni potevano essere considerate soddisfacenti e non si avvertiva la necessità di affrontare direttamente il problema dinamico delle singole collisioni.
Gli importanti progressi realizzati negli ultimi decenni, legati in gran parte all'avvento di nuove tecniche sperimentali (raggi molecolari, spettroscopie risolte in tempi molto brevi, ecc., Tavv. I e II), hanno reso possibile lo studio della dinamica delle singole collisioni. In casi favorevoli, questi dati permettono di ottenere a ritroso informazioni sulle forze d'interazione, fornendo così un ponte tra eventi osservati e leggi dinamiche fondamentali. Il programma attuale della cinetica chimica sperimentale, dunque, consiste nel fornire dati sempre più accurati sulla dinamica dei processi elementari, mentre dalla teoria ci si attende modelli interpretativi e metodi di approssimazione sempre più raffinati. Occorre sottolineare che la teoria delle collisioni costituisce un campo estremamente vasto della fisica teorica: le tecniche collisionali, infatti, se rappresentano un recente strumento d'indagine nella cinetica chimica, costituiscono invece il metodo essenziale nella fisica nucleare e in quella delle particelle. In effetti, ogni informazione sulla struttura della materia e sulle sue trasformazioni è il risultato di collisioni tra particelle elementari.
Anche se la descrizione dei processi molecolari è riconducibile alla meccanica quantistica, per molti problemi d'interesse può essere sufficiente muoversi nell'ambito del limite classico. Una metodologia spesso rilevante per la dinamica chimica consiste, quindi, nell'impostare in maniera classica un determinato problema e nell'introdurre successivamente un formalismo quantomeccanico. In questo modo, si sono sviluppate teorie semiclassiche delle collisioni, in cui le particelle seguono traiettorie classiche e manifestano proprietà quantistiche (ondulatorie) soltanto in alcune regioni critiche del loro cammino. In questo ambito, è possibile descrivere i fenomeni molecolari con un formalismo analogo a quello dell'ottica ondulatoria. Tale analogia si riflette anche nel linguaggio: per esempio, effetti d'interferenza quantomeccanica prendono il nome di aureola o arcobaleno.
Dinamica delle reazioni
Il problema fondamentale della dinamica chimica consiste nella ricerca di relazioni tra le forze che si esercitano a livello molecolare e le grandezze osservabili (per es., le sezioni d'urto). Questo procedimento, tipicamente riduzionistico, in molti casi è a senso unico. Infatti, il cosiddetto problema dell'inversione, consistente nel risalire dalle sezioni d'urto alle forze di interazione, è risolubile soltanto in situazioni particolarmente semplici. Più spesso, è necessario costruire un modello per le interazioni che sia, da un lato, sufficientemente realistico e, dall'altro, abbastanza trattabile, e quindi impostare il problema dinamico relativo, confrontando infine i risultati con quelli degli esperimenti. L'interazione tra le particelle dipende da tanti termini quante sono le interazioni tra ognuno degli elettroni e dei nuclei che le compongono e tutti gli altri elettroni e nuclei. Le superfici di energia potenziale avranno una dimensionalità pari a tre volte il numero totale di particelle (tre coordinate per ogni nucleo e tre per ogni elettrone), e per questa ragione possono essere descritte da funzioni assai complicate. È quindi importante averne, con riferimento a un processo specifico, una visualizzazione appropriata e una rappresentazione matematica in funzione di un numero ridotto di coordinate ritenute cruciali per la descrizione della dinamica delle reazioni. Comunemente, si adotta la separazione di Born-Oppenheimer, che sfrutta l'enorme differenza tra le masse dei nuclei e quelle degli elettroni, per cui si può descrivere il moto (lento) dei nuclei nel campo medio prodotto dal moto veloce degli elettroni. Si ottengono così le superfici di energia potenziale in funzione delle sole coordinate nucleari. In generale, è ben caratterizzabile la forma delle superfici di energia potenziale per le configurazioni corrispondenti ai reagenti e ai prodotti, cioè alle situazioni di partenza e di arrivo delle reazioni. Infatti, reagenti e prodotti sono in genere sostanze sufficientemente stabili da poter essere studiate con le tecniche convenzionali dell'analisi chimica e della spettroscopia atomica e molecolare. Assai meno note sono le configurazioni corrispondenti agli stadi intermedi delle reazioni (complessi intermedi o stati di transizione).
Per quanto riguarda gli stadi intermedi e la caratterizzazione dei cammini di reazione tramite i quali i reagenti danno origine ai prodotti, è fondamentale il contributo della chimica teorica moderna, che permette una trattazione quantistica della struttura elettronica delle molecole e consente di individuare accuratamente le modificazioni geometriche intervenute a livello molecolare nel corso delle reazioni chimiche e di classificare le simmetrie dominanti. Tuttavia, dato che le previsioni della chimica teorica non hanno ancora raggiunto una sufficiente accuratezza per quanto riguarda le caratteristiche energetiche fondamentali della dinamica delle reazioni (esotermicità o endotermicità, energia di attivazione), per queste grandezze si fa ricorso anche a modelli totalmente o parzialmente empirici. A partire dagli anni Sessanta del Novecento, sono stati compiuti grandi progressi in questa direzione. La possibilità di trattare accuratamente il dettaglio della dinamica di una reazione chimica elementare come problema quantomeccanico dell'interazione fra tre corpi è emersa invece negli anni Settanta, grazie all'avvento di strumenti di calcolo sempre più potenti. Dove sono stati possibili confronti, si è arrivati alla conclusione che le tecniche basate esclusivamente sulla meccanica classica, che quindi trascurano i tipici effetti quantomeccanici quali l'interferenza, l'effetto tunnel e le risonanze, forniscono in generale una stima accettabile per le costanti di velocità, almeno per reazioni non coinvolgenti gli atomi di idrogeno, mentre è necessario un approccio esplicitamente quantomeccanico per la previsione accurata delle sezioni d'urto. In base a queste considerazioni, sono stati sviluppati procedimenti intermedi, generalmente designati come semiclassici, in cui l'evoluzione dinamica sulle superfici di energia potenziale è descritta come moto essenzialmente classico dei nuclei, ma gli effetti quantistici sono introdotti tenendo conto esplicitamente del comportamento ondulatorio nel regime di lunghezze d'onda piccole rispetto alle dimensioni molecolari. Per quanto riguarda la trattazione di problemi più complicati di quelli delle reazioni a tre centri, l'uso di tecniche classiche, semiclassiche o statistiche si rende necessario a causa della difficoltà di affrontare direttamente la soluzione delle equazioni della meccanica quantistica.
Lo stato di transizione
È opportuno approfondire la teoria dello stato di transizione, che ha lo scopo di formulare in termini quantistici il procedere di una reazione attraverso uno o più stati intermedi. La teoria è anche nota come teoria del complesso attivato, o teoria delle velocità assolute di reazione, e il fatto stesso che le si attribuiscano molti nomi e che abbia avuto varie formulazioni alternative testimonia come lo stato di transizione sia difficile non solo da caratterizzare sperimentalmente, ma anche da mettere a fuoco dal punto di vista teorico. Henry Eyring, uno dei fondatori della teoria, nel 1934 ne propose una versione termodinamica tuttora utilizzata. In essa, allo stato di transizione sono attribuiti valori delle funzioni termodinamiche (entalpia ed entropia, e quindi energia libera), alla stregua di un ben definito stato di equilibrio termodinamico. Poiché le tecniche di calcolo quantomeccanico dell'epoca non erano sufficientemente precise da consentire un'accurata caratterizzazione dello stato di transizione, s'ipotizzava che a questo corrispondesse un minimo energetico nella superficie di energia potenziale, detto il lago di Eyring. Incidentalmente, è interessante far notare che la dinamica di una reazione chimica è spesso descritta con un linguaggio pittoresco; per esempio, nel rappresentare l'evoluzione del sistema reattivo sulle superfici di energia potenziale, introdotte all'inizio degli anni Trenta da Eugene Wigner e Michael Polanyi, si parla di valle dei reagenti e dei prodotti, e si dice che la reazione avviene per superamento di una sella o di un crinale.
Le ricerche teoriche successive al lavoro di Eyring indicarono come erronea l'idea del lago, vale a dire di una buca di potenziale sulla sella, dove è localizzato lo stato di transizione (o complesso intermedio). Le ricerche recenti sulla caratterizzazione, anche sperimentale, dello stato di transizione sono state inquadrate in una prospettiva storica da Zewail nel libro The chemical bond: structure and dynamics (1992). Negli ultimi anni, grazie soprattutto ai lavori di Zewail, si è aperta la strada per rendere possibile l'osservabilità dello stato di transizione (Tav. II). Questa prospettiva si è concretizzata con lo sviluppo delle tecniche basate sulla spettroscopia laser ultraveloce e sui fasci molecolari. Grazie a queste tecniche è possibile ottenere informazioni sperimentali su scale di tempi di interazione brevissimi (dell'ordine dei femtosecondi) e dunque seguire eventi singoli nella reazione.
Viste le difficoltà inerenti alla caratterizzazione sperimentale dello stato di transizione, un ruolo decisivo è giocato dalla chimica teorica che, sin dagli anni Trenta, si è posta il problema del calcolo delle sue proprietà, anche se con risultati spesso contraddittori. Possibili equivoci e ingiustificati ottimismi sono stati successivamente ridimensionati dall'affinamento delle tecniche computazionali e dal potenziamento degli strumenti di calcolo. I notevoli ostacoli incontrati dagli approcci della chimica quantistica, spesso indicati genericamente come ab initio, sono fortemente dipendenti dalle capacità di memoria e di potenza di calcolo dei computer, oltre che, ovviamente, dall'adeguatezza del modello e dalle tecniche di programmazione. Il chimico, ormai, ha a disposizione strumenti di calcolo molto elaborati. Nel 1998, il premio Nobel per la chimica è stato assegnato a John A. Pople proprio per il suo contributo in questa direzione. Il riconoscimento è stato condiviso con Walter Kohn, premiato per aver contribuito alla ricerca di vie di calcolo alternative a quelle tradizionali della chimica quantistica (in particolare, con il metodo del funzionale della densità). Tuttavia, a causa della pesantezza dei calcoli, non è sorprendente che continuino a essere proposte e sviluppate altre tecniche, anche di natura semiempirica.
La descrizione di aspetti complessi della chimica ‒ come, per esempio, la relazione tra struttura e reattività, oppure il comportamento in condizioni estreme di pressione e di temperatura e così via ‒ fa tuttora ampio ricorso a correlazioni empiriche che, da un lato, tentano di compattare un insieme molto ampio di eventi sperimentali entro schemi semplici e con una drastica riduzione del numero di variabili, dall'altro lato, si propongono come agile strumento di previsione di fenomeni non ancora osservati e quale stimolo per ulteriori misurazioni sperimentali.
Dinamica molecolare dei sistemi complessi
Nel corso degli ultimi decenni si è realizzato uno sviluppo molto significativo nello studio della dinamica molecolare dei sistemi che coinvolgono fasi condensate, utilizzando tecniche sperimentali e teorico-interpretative basate su principî simili a quelli esposti sinora con riferimento alla sola fase gassosa. Le tecniche dei fasci molecolari e della spettroscopia ultraveloce sono risultate particolarmente utili per la caratterizzazione della dinamica dei processi reattivi che avvengono alle interfasi, così come per quella dei processi che coinvolgono aggregati molecolari (cluster) di dimensioni sempre crescenti. La reattività in sistemi coinvolgenti superfici e aggregati è oggetto di intensi studi teorici; nelle tecniche utilizzate, un ruolo fondamentale è svolto dalle simulazioni che fanno esplicito riferimento alla meccanica classica come approssimazione al comportamento quantistico, che è in generale più difficile da descrivere. Si prevede che nei prossimi anni queste simulazioni potranno affinarsi utilizzando sia descrizioni più accurate delle interazioni intermolecolari (per es., ottenute da esperimenti come quelli illustrati nella Tav. I), sia trattazioni in cui la meccanica classica e quella quantistica siano opportunamente combinate (per es., il metodo simulativo Car-Parrinello). Ulteriori sviluppi riguardano i settori fondamentali della chimica delle reazioni nello stato di soluzione, soprattutto acquosa, e della reattività di sistemi intrappolati in matrici solide.
La modellizzazione di sistemi macroscopici come insiemi di processi microscopici è uno dei principali campi di attività dell'attuale ricerca in chimica ed è importante che si faccia almeno un accenno ai nuovi problemi metodologici. Un punto fondamentale riguarda la non linearità delle equazioni fenomenologiche, che descrivono le variazioni nel tempo delle concentrazioni delle specie coinvolte in un processo (diminuzione dei reagenti, aumento dei prodotti). Tale non linearità viene discussa nell'ambito delle problematiche associate al cosiddetto caos chimico che emerge nella modellistica dei sistemi complessi. Ulteriori aspetti di particolare interesse riguardano il ruolo delle tecniche semiclassiche (o teorie asintotiche) nella descrizione quantistica di sistemi che classicamente si presentano caotici, laddove la trattazione esatta, basata sulla meccanica quantistica (che è essenzialmente una teoria lineare), non prevede l'emergenza di tale fenomeno.
Il tema del rapporto tra mondo microscopico e mondo macroscopico, strettamente legato a quello tra meccanica classica e meccanica quantistica (e anche della linearità e della non linearità, del determinismo e dell'indeterminazione, della complessità e del caos), assume notevole importanza e trova ulteriore sviluppo nelle ricerche di meccanica statistica, che hanno lo scopo di costituire la base microscopica della termodinamica chimica. Mentre la teoria della velocità delle reazioni, come è stata sviluppata da Arrhenius, nella sua interpretazione in termini dei modelli collisionali della teoria cinetica dei gas, ha sempre fatto ricorso alla realtà microscopica nell'interpretazione dell'energia di attivazione e del fattore pre-esponenziale, l'approccio termodinamico di Eyring è il più importante esempio del tentativo di mantenere la descrizione della realtà chimica entro uno schema di linguaggio ‒ quello della termodinamica ‒ specifico della realtà macroscopica.
Questo è anche l'ambito entro cui si sviluppano le ricerche di Ilya Prigogine, premio Nobel per la chimica nel 1977. Si tratta di un'alternativa, diremmo fenomenologica, rispetto al progetto riduzionistico della meccanica statistica, mirante a interpretare come opportune medie su comportamenti microscopici le quantità che compaiono nelle formulazioni termodinamiche. La dinamica molecolare contribuisce a tale progetto mediante una modellistica che fa amplissimo uso di simulazioni computazionali. Lo scenario della ricerca in questo campo si è andato talmente modificando da trasformare in modo profondo il ruolo e gli strumenti d'indagine del chimico moderno rispetto alle problematiche della scienza dei materiali, offrendo una serie di spunti per ulteriori indagini, che potranno condurre ad acquisizioni di rilevante interesse non soltanto per la ricerca chimico-fisica più avanzata, ma anche, più in generale, dal punto di vista delle prospettive di progresso delle scienze della natura.
bibliografia
Aquilanti, Mele 1992: Aquilanti, Vincenzo - Mele, Antonio, Cinetica chimica, in: Enciclopedia delle scienze fisiche, Roma, Istituto della Enciclopedia Italiana, 1992, I, pp. 603-608.
Aquilanti, Volpi 1998: Aquilanti, Vincenzo - Volpi, Gian Gualberto, Dinamica delle reazioni chimiche, in: Enciclopedia del Novecento, Roma, Istituto della Enciclopedia Italiana, 1998, XI, suppl. II, pp. 573-582.
Bonino 1931: Bonino, Giovanni Battista, Cinetica chimica, in: Enciclopedia Italiana di Scienze, Lettere ed Arti, Roma, Istituto della Enciclopedia Italiana, 1931, X, pp. 358-367.
Herschbach 1993: Herschbach, Dudley R., Molecular dynamics of elementary chemical reactions, in: Nobel lectures in chemistry: 1981-1990, edited by Bo G. Malmstrom, Singapore, World Scientific, 1993, pp. 265-314.
Lee 1993: Lee, Yuan T., Molecular beam studies of elementary chemical processes, in: Nobel lectures in chemistry: 1981-1990, edited by Bo G. Malmstrom, Singapore, World Scientific, 1993, pp. 320-354.
Levi 1993: Levi, Andrea Claudio, Fasci molecolari, in: Enciclopedia delle scienze fisiche, Roma, Istituto della Enciclopedia Italiana, 1993, II, pp. 522-525.
Levine, Bernstein 1987: Levine, Raphael D. - Bernstein, Richard B., Molecular reaction dynamics and chemical reactivity, New York-Oxford, Oxford University Press, 1987.
Polanyi 1993: Polanyi, John C., Some concepts in reaction dynamics, in: Nobel lectures in chemistry: 1981-1990, edited by Bo G. Malmstrom, Singapore, World Scientific, 1993, pp. 359-407.
Rolla 1948: Rolla, Mario, Cinetica chimica, in: Enciclopedia Italiana di Scienze, Lettere ed Arti, Appendice II, Roma, Istituto della Enciclopedia Italiana, 1948, pp. 614-617.
Zewail 1992: Zewail, Ahmed H., The chemical bond: structure and dynamics, San Diego, Academic Press, 1992.
Zewail 2000: Zewail, Ahmed H., Femtochemistry: atomic-scale dynamics of the chemical bond, "Journal of physical chemistry A", 104, 2000, pp. 5660-5694.
Tavola I
Lo studio della dinamica delle reazioni chimiche: le tecniche collisionali
Nello studio della dinamica molecolare, le grandezze misurabili, associate alla probabilità di un evento reattivo, sono le sezioni d’urto, il cui significato è illustrato nella figura, con riferimento a due possibili configurazioni sperimentali. Le due apparecchiature mostrate consentono di misurare, rispettivamente, sezioni d’urto integrali (in alto) e sezioni d’urto differenziali (configurazione a fasci molecolari incrociati, in basso). Nel primo caso è possibile ottenere l’andamento della sezione d’urto integrale in funzione della velocità di collisione, nel quale sono evidenziabili ad alte velocità l’effetto della repulsione molecolare e a basse velocità quello dell’attrazione, su cui si sovrappone una interferenza di natura quantomeccanica detta di aureola. Queste misure richiedono intensità notevoli di fasci molecolari.
Nell’esempio nella figura si tratta di fasci supersonici, collimati da skimmer (scrematori, schiumatori) e selezionati in velocità, ulteriormente analizzati nello stato magnetico e rivelati mediante spettrometria di massa a quadrupolo. La misura delle sezioni d’urto differenziali (in basso, nella figura) permette di costruire grafici di tale grandezza in funzione dell’angolo di diffusione, nei quali sono osservabili alcuni effetti interferenziali come, per esempio, il fenomeno dell’arcobaleno molecolare. Da tali grafici è possibile ricostruire, nei casi più favorevoli, gli aspetti essenziali dell’andamento del potenziale intermolecolare (grafico al centro).
Tavola II
Lo studio della dinamica delle reazioni chimiche: le tecniche spettroscopiche
Le tecniche spettroscopiche sono usate da vari decenni per lo studio della dinamica delle reazioni chimiche. Tuttavia, esse hanno subito un particolare impulso con lo sviluppo e con la crescente disponibilità di sorgenti di radiazione laser. Tra le metodiche di più recente introduzione, molto importante è la spettroscopia basata sulla produzione di impulsi di radiazione laser ultrabrevi, dell’ordine di grandezza dei femtosecondi (1 fs = 10-15 s). Un esempio di apparecchiatura per spettroscopia laser ultraveloce è illustrato nella figura. La reazione viene innescata con un impulso laser di pompaggio, che fissa il tempo zero della reazione, e la sua evoluzione viene seguita mediante impulsi di sonda, che eccitano o ionizzano selettivamente i reagenti, i prodotti o lo stato di transizione. L’evoluzione temporale della reazione, a energia e stati iniziali e finali definiti, può essere seguita variando i tempi di ritardo tra i due impulsi. Nel grafico qui riportato viene mostrata l’evoluzione temporale del segnale LIF in funzione del ritardo dell’impulso di sonda rispetto a quello di pompaggio, per un processo di decomposizione di una molecola triatomica che fornisce come prodotti una specie monoatomica e una biatomica. Le diverse curve si riferiscono a differenti valori di lunghezza d’onda dell’impulso di sonda. Il processo di dissociazione è rappresentato nel diagramma in basso a sinistra, dove sono riportate anche, schematicamente, le superfici di energia potenziale. La risoluzione temporale di questi metodi è ormai giunta all’ordine
di pochi femtosecondi, ed è dunque dello stesso ordine di grandezza del tempo di vita degli stati di transizione. Dopo oltre cinquant’anni dalla teorizzazione dello stato di transizione (v. par. 4), si è così aperta la strada alla sua osservazione diretta per via sperimentale. L’enorme rilevanza concettuale che deriva dall’acquisizione di questi nuovi metodi di indagine è stata sancita, tra l’altro, dall’assegnazione del premio Nobel per la chimica nel 1999 ad Ahmed H. Zewail, che ha fornito un contributo particolarmente significativo al loro sviluppo.