
demenza
demenza
Il termine demenza definisce una compromissione stabile delle funzioni cerebrali superiori acquisite ed esclude tutti gli stati di insufficienza mentale transitoria o secondaria a sviluppo deficitario. Il concetto di demenza implica un’incapacità da parte dell’individuo affetto di rispondere alle proprie esigenze quotidiane. A essa si associano deficit cognitivi misurabili, alterazioni dello stato emozionale e disturbi comportamentali. Esistono numerose condizioni patologiche che possono indurre uno stato di demenza, di cui la malattia di Alzheimer rappresenta la forma più comune. Essa, assieme ad altre forme più rare, è considerata a eziologia ignota e si sviluppa attraverso meccanismi neurodegenerativi. Al contrario, esistono forme di demenza secondaria, che riconoscono una causa specifica (per es., la demenza cerebrovascolare). Sono oggi disponibili strumenti per la diagnosi precoce delle diverse forme di demenza e terapie specifiche di tipo sia farmacologico che comportamentale. [➔ Alzheimer, malattia di; funzioni cerebrali superiori; invecchiamento cerebrale; neurodegenerazione; Parkinson, malattia di] Il termine deriva dal latino dementia («privo di mente») e definisce una compromissione globale delle funzioni cerebrali superiori, in assenza di disturbi della vigilanza. Le funzioni superiori includono abilità cognitive come la memoria, l’attenzione, le capacità esecutive (pianificazione, organizzazione) e visuospaziali, le prassie (abilità di eseguire gesti in assenza di deficit motori), il linguaggio e l’elaborazione degli stati emozionali. Lo stato di d. che, a seconda della condizione clinica sottostante, si può associare a diversi profili cognitivi deficitari, implica come condizione necessaria l’incapacità da parte del soggetto affetto di far fronte alle richieste della vita quotidiana, di conservare un comportamento adeguato alle circostanze, di controllare i propri stati emotivi. Per definizione, la condizione di d. implica la perdita stabile di abilità precedentemente acquisite, in opposizione a quella di oligofrenia, che definisce invece una carenza delle stesse abilità a causa di uno sviluppo deficitario. Il concetto di d. va considerato alla stregua di un’insufficienza cronica d’organo (il cervello) e va distinto dalle insufficienze acute e transitorie. La definizione operativa contenuta nel DSM-IV (➔) dell’American Psychiatric Association prevede l’accertamento di un deficit di memoria in associazione a uno o più dei seguenti deficit cognitivi: afasia, aprassia, agnosia, compromissione delle funzioni esecutive. L’insieme di questi deficit deve produrre una inadeguatezza del funzionamento sociale o lavorativo dell’individuo affetto, e non deve essere riconducibile allo stato confusionale acuto o alle patologie psichiatriche. Nel passato si usava anche il termine amenza per definire una grave sindrome psichica secondaria ad alcune condizioni cliniche (malattie infettive, intossicazioni, lesioni cerebrali o traumi emozionali) e caratterizzata da un’intensa agitazione psicomotoria, incapacità di orientamento spaziotemporale, ansia, deficit di memoria, ideazioni deliranti.
Nosologia delle sindromi demenziali
Le d. possono essere classificate in forme neurodegenerative primarie e secondarie. Le principali forme di d. degenerativa primaria includono la malattia di Alzheimer (MA), la d. a corpi di Lewy (dementia with Lewy bodies, DLB), la d. fronto-temporale (FTD), la d. associata a malattia di Parkinson (PDD) e la degenerazione cortico-basale (CBD). Le principali forme secondarie includono la d. vascolare (VAD) e l’idrocefalo normoteso (NPH), che più frequentemente di altre pongono difficoltà diagnostiche differenziali con le forme degenerative primarie. Nel capitolo delle d. secondarie vanno inoltre menzionate le forme post-traumatiche (PTD), le d. da atrofia cerebrale paracarcinomatosa, le d. da ipossia cerebrale protratta (per es., insufficienza emodinamica acuta; intossicazione da ossido di carbonio), le d. distiroidee e carenziali, le d. collagenopatiche, le d. post-infettive (incluse quelle da malattia prionica), le pseudodemenze. Queste ultime possono essere di natura iatrogena, metabolica, o secondarie a malattia psichiatrica.
Epidemiologia
L’incidenza (numero di nuovi casi per anno) di d. stimata su scala mondiale varia da un minimo di 3,5 casi ogni 1.000 abitanti in Africa, a un massimo di 10 casi per mille abitanti in America Settentrionale, con un’incidenza media di 7,5 casi per mille. La prevalenza (numero di casi esistenti) mondiale media è di 3,5 casi ogni 100 persone. Si può stimare che, a livello planetario, nel 2020 saranno 42 milioni gli individui dementi e che questo numero è destinato ad aumentare a 81 milioni nel 2040. La variabilità epidemiologica riscontrata nelle diverse aree geografiche è da ascriversi, oltre a fattori ambientali, genetici e altri che è impossibile quantificare, alla proporzione relativa differente di individui anziani nelle diverse popolazioni. La MA (principale forma di d.) riconosce infatti nell’invecchiamento il proprio fattore di rischio maggiore. Per quanto riguarda i diversi tipi di d., secondo un recente studio condotto dalla Florida Brain Bank, (che possiede dati epidemiologici sulle malattie cerebrali) la MA costituisce la forma più rappresentata (Frequenza Relativa, FR: 42÷77%), seguita dalla DLB (FR: 8÷26%), dalla VaD (FR: 3÷18%) e dalla FTD (FR: 4÷5%).
Demenze neurodegenerative primarie
Le forme neurodegenerative primarie esordiscono caratteristicamente in età senile (oltre i 60 anni di età, ma possono manifestarsi anche più precocemente). La loro eziologia è sconosciuta e, nonostante i recenti progressi, i meccanismi fisiopatologici sottostanti risultano solo parzialmente compresi. Nella pratica clinica, la diagnosi differenziale delle d. degenerative si basa su criteri clinici, evidenze psicometriche, marcatori neuroaradiologici e liquorali. Tipicamente, viene formulata una diagnosi probabile di una determinata forma di d. neurodegenerativa in accordo a criteri diagnostici specifici e convalidati. Essi prevedono dapprima l’esclusione di tutte le possibili forme secondarie, alcune delle quali suscettibili di trattamento specifico. Il quadro clinico, neuropsicologico e strumentale guida pertanto la diagnosi verso la forma più probabile di d. primaria: la certezza diagnostica si ottiene infatti unicamente sulla base del riscontro autoptico. Col progredire delle conoscenze sta divenendo sempre più importante un’esatta e precoce formulazione diagnostica. Essa ha infatti implicazioni non solo di tipo speculativo, ma anche terapeutico.
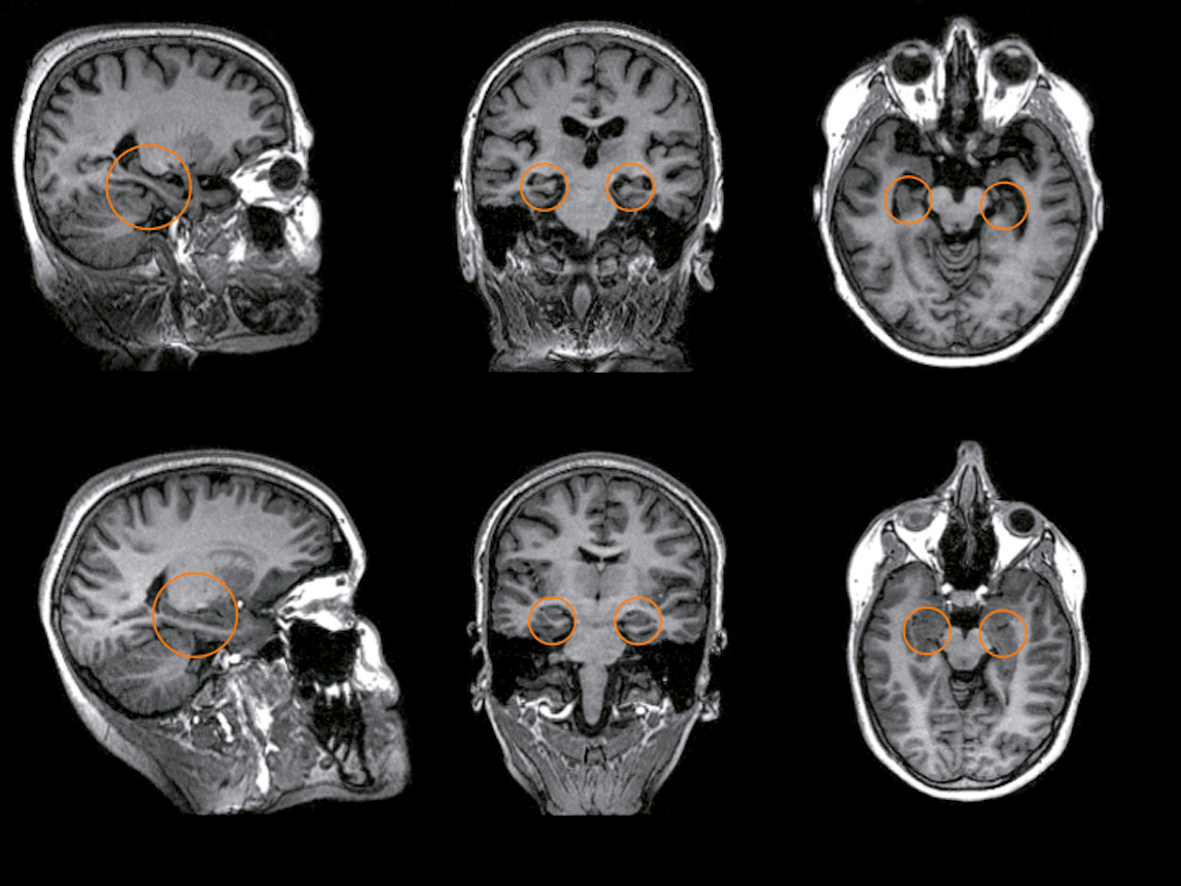
Malattia di Alzheimer. Tipicamente, si caratterizza per la comparsa di deficit di memoria a carattere ingravescente, a cui, successivamente, si aggiungono deficit di altri domini cognitivi (attenzione, prassia, abilità esecutive, ecc.) sino alla conversione a stadio di d. conclamata. La MA è per larga parte da considerarsi una malattia sporadica. Tuttavia, in un numero ristretto di casi (tipicamente a esordio in età presenile), è stata riconosciuta una variante familiare di malattia, strettamente associata a specifiche mutazioni genetiche (per es., gene precursore della β-amiloide, gene della presenilina 1 e 2). Nella sua forma sporadica, la MA è stata anche associata a una specifica forma allelica (e4) del gene per la apolipoproteina E, ancorché essa costituisca solo un fattore predisponente.
Demenza a corpi di Lewy e demenza associata a malattia di Parkinson. La DLB è stata riconosciuta come la seconda più comune forma di d., sebbene, nella pratica clinica, essa rimanga largamente diagnosticata in maniera non corretta. Clinicamente si caratterizza per la comparsa di decadimento cognitivo con un prevalente coinvolgimento delle funzioni attentive, esecutive e visuospaziali e un relativo risparmio delle funzioni mnesiche (almeno nelle fasi più iniziali). Agli aspetti cognitivi si associano altri sintomi (core features, sintomi cardinali) indispensabili per la diagnosi, quali la presenza di una sindrome parkinsoniana (rigido-acinetica), di fluttuazioni cognitive e di allucinazioni visive ricorrenti (tipicamente ben strutturate). Ulteriori elementi che supportano la diagnosi di DLB (suggestive features, sintomi di supporto) sono la presenza di disturbi comportamentali, una grave ipersensibilità alle terapie neurolettiche e un ridotto accumulo a livello del nucleo striato (posto alla base dell’encefalo) del trasportatore della dopammina, che viene valutato mediante SPECT (tomografia a emissione di singolo fotone). Le tecniche di neuroimmagine mostrano un’atrofia delle strutture temporali mediali, meno rilevante rispetto a quella riscontrabile in pazienti con MA. La PDD che esordisce con disturbi motori si associa frequentemente a sviluppo di d. con caratteristiche analoghe a quelle della DLB. I rapporti tra le due malattie sono ancora oggetto di discussione scientifica; da un punto di vista patologico, sia la DLB sia la PDD si caratterizzano infatti per la presenza dei cosiddetti corpi di Lewy, che nel primo caso presentano una distribuzione più diffusa (sia corticale sia sottocorticale), mentre nel secondo risultano più confinati alle strutture sottocorticali.
Demenza frontotemporale. La FTD (detta anche d. frontotemporale di Pick) costituisce un gruppo di malattie (con possibile esordio anche in età presenile) che, in stadio tardivo o intermedio, possono risultare clinicamente indistinguibili dalla MA. Nelle fasi precoci, al contrario, esse si caratterizzano per la predominanza di disturbi comportamentali, affettivi e del linguaggio, con deficit di memoria modesti o addirittura assenti. Queste malattie vengono generalmente distinte nelle seguenti sottocategorie: variante frontale (50% di tutti i casi di FTD), dominata da disturbi comportamentali; afasia progressiva non fluente, dominata da deficit delle funzioni linguistiche; d. semantica, dominata da deficit delle conoscenze concettuali. Clinicamente i pazienti affetti dalla variante frontale esordiscono con modificazioni del loro comportamento sociale (inerzia, abulia, disinibizione, disturbi attentivi); i test psicometrici evidenziano deficit prevalenti alle prove di ragionamento astratto e di categorizzazione. I pazienti affetti da afasia progressiva non fluente esordiscono invece con difficoltà nel reperire parole (anomia) e con marcata diminuzione della fluenza verbale. In alcuni casi l’insorgenza del decadimento cognitivo generalizzato può seguire anche di diversi anni i disturbi iniziali del linguaggio. Nella forma semantica, infine, i pazienti mostrano difficoltà di denominazione di oggetti, pur mantenendo una buona fluenza verbale, ancorché ricca di circumlocuzioni e parafasie semantiche. A un esame neuropsicologico accurato può essere dimostrata una conoscenza conservata degli stimoli presentati (uso, funzione, forma, ecc.) con difficoltà selettiva all’accesso lessicale. La risonanza magnetica può mostrare atrofia cerebrale regionale coerente con i disturbi neuropsicologici.
Degenerazione corticobasale. La CBD è una forma di d. per certi versi accomunabile alla FTD. Caratteristicamente esordisce con sintomi motori (della serie parkinsoniana) insidiosi e progressivi e con distribuzione asimmetrica. Abbastanza caratteristica è la perdita di sensibilità complesse (per es., alterazioni dello schema corporeo), prevalenti dal lato opposto all’emisfero cerebrale più colpito, con comparsa del cosiddetto arto alieno (incapacità a riconoscere come proprio un arto). La sindrome neuropsichiatrica si caratterizza per d. frontale e sottocorticale, apatia, irritabilità, agitazione, deliri, disinibizione, tratti ossessivo-compulsivi. Di particolare aiuto diagnostico il riscontro con RM di un’atrofia emisferica asimmetrica. Non vi sono alterazioni patologiche caratteristiche.
Paralisi sovranucleare progressiva. Si tratta di un’altra forma di d. degenerativa associata a parkinsonismo. Si caratterizza per il coinvolgimento di strutture sottocorticali, con deficit frontali e manifestazioni caratteristiche come la paralisi verticale di sguardo, l’instabilità posturale e disturbi cognitivo-comportamentali. La sua differenziazione con la PDD può risultare difficoltosa.
Principali demenze secondarie
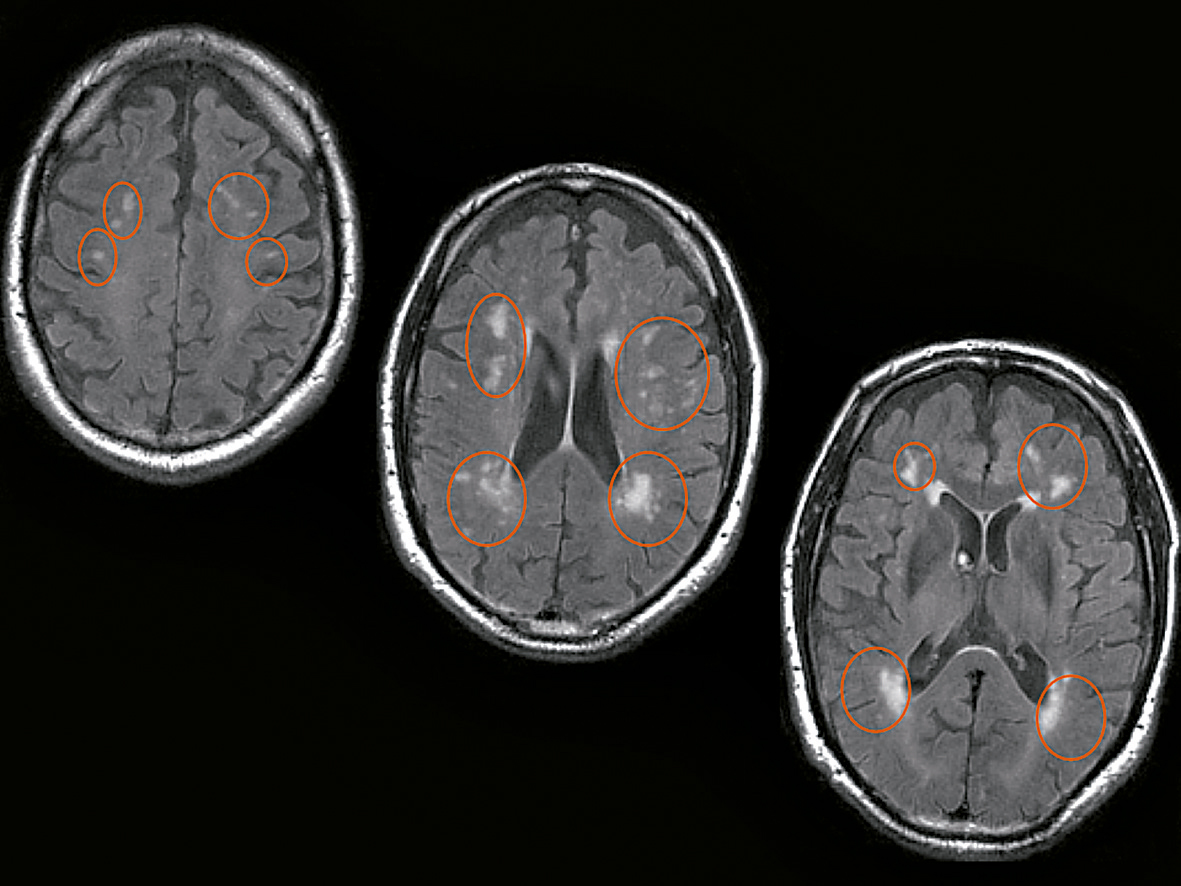
Demenza vascolare. Si raggruppano in questo capitolo una serie di quadri clinici e fisiopatologici estremamente diversi tra di loro, ma accomunati dalla genesi vascolare del danno cerebrale. La loro differenziazione si basa su elementi anamnestici, clinici e su esami per immagini dell’encefalo, in particolare la RM, particolarmente sensibile alle lesioni vascolari. La VaD multinfartuale è causata da ictus maggiori ricorrenti, che si associano a un decadimento cognitivo cosiddetto a gradini, dove ognuno di essi corrisponde a un nuovo evento ischemico. Questa forma trova nelle condizioni cardio-emboliche e nell’aterosclerosi delle grosse arterie cerebrali le sue maggiori cause predisponenti. La VaD sottocorticale è invece dovuta a occlusione dei piccoli vasi. La sua causa più comune è rappresentata dall’arteriosclerosi su base ipertensiva. Il decorso clinico presenta un decadimento cognitivo progressivo che, a differenza dalla MA, non è dominato dai disturbi di memoria. La diagnosi differenziale fra VaD sottocorticale e MA può risultare tuttavia particolarmente difficile, specialmente in quei casi in cui le due forme di d. coesistono (d. mista). La VaD da singolo infarto strategico si caratterizza per l’insorgenza acuta di una lesione vascolare in un territorio critico per le funzioni cognitive, come il lobo temporale mediale, il lobo frontale, il giro angolare, la testa del nucleo caudato, il talamo, il ginocchio della capsula interna. A seconda della sede coinvolta può manifestarsi con sintomi cognitivi puri o in associazione con altri deficit neurologici focali, come una paralisi motoria. Come per la MA, anche per le d. vascolari è stato introdotto il concetto di diagnosi preclinica mediante la definizione del deficit cognitivo vascolare (VCI, Vascular Cognitive Impairment). Idrocefalo normoteso. Questa condizione clinica è dovuta a un incremento della pressione liquorale intraventricolare, con riassorbimento del liquor cefalorachidiano a livello del parenchima cerebrale. Clinicamente si caratterizza (nella sua espressione completa) per la presenza di una triade di sintomi, costituita da: deficit cognitivi prevalentemente frontali, disturbo della deambulazione (a piccoli passi), disturbi minzionali. La diagnosi si basa sul monitoraggio invasivo della pressione liquorale o sullo studio della dinamica liquorale mediante cisternografia radioisotopica. Il riconoscimento di questa malattia è di cardinale importanza, in quanto essa è passibile di trattamento chirurgico specifico.
Malattie prioniche. Si tratta di una famiglia di rarissime condizioni patologiche, dementigene e fatali, che necessitano di essere tempestivamente riconosciute. La più nota è la malattia di Creutzfeldt-Jakob, dovuta a infezione da parte di ceppi diversi di una proteina prionica (➔ prione) patologica (PrPSc). In caso di sospetto diagnostico, vanno tempestivamente messi in atto una serie di accertamenti indispensabili (elettroencefalogramma, positivo nell’80% dei casi; RM encefalo, esame liquorale). Esiste infatti un rischio di trasmissione per esposizione di tessuti contaminati, come nel caso, per es., del trapianto di cornea.
Terapia delle demenze
Esistono diversi livelli di intervento, a seconda del tipo e della gravità della condizione dementigena considerata. Essi si dividono in prevenzione primaria, prevenzione secondaria e trattamento della d. clinicamente manifesta. Relativamente all’ultimo livello, sono oggi disponibili i farmaci anticolinesterasici (donepezil, rivastigmina e galantamina), i quali aumentano la biodisponibilità di acetilcolina a livello delle sinapsi nervose e risultano efficaci nel migliorare, sebbene per periodi limitati di tempo, i deficit cognitivi della MA. Essi vengono utilizzati con successo anche nel trattamento della DLB, riducendo in modo significativo le allucinazioni visive e le fluttuazioni cognitive. La memantina, usata nella MA, riduce l’effetto citotossico dell’acido glutammico e, potenzialmente, gli aspetti neurodegenerativi. Altri promettenti approcci terapeutici per le d. degenerative sono attualmente (2010) in fase di sperimentazione (per es., il vaccino anti �β-amiloide). Le d. secondarie vanno trattate in base alla loro causa primaria: terapia antiaggregante o anticoagulante nelle VaD; derivazione ventricolo-peritoneale nell’NPH; correzione dei deficit in tutte le cause metaboliche e carenziali. Singoli sintomi comportamentali possono essere trattati con antipsicotici, antidepressivi, ansiolitici e anticonvulsivanti. A complemento delle terapia farmacologica, esistono inoltre strategie comportamentali. In partic., le tecniche di attivazione cognitiva aspecifica vengono utilizzate per stimolare capacità residue e migliorare l’esecuzione di compiti quotidiani nei pazienti con demenza. È infine rilevante il supporto (informativo e psicologico) rivolto ai caregivers ossia a coloro che accudiscono i pazienti. Carlo Caltagirone