
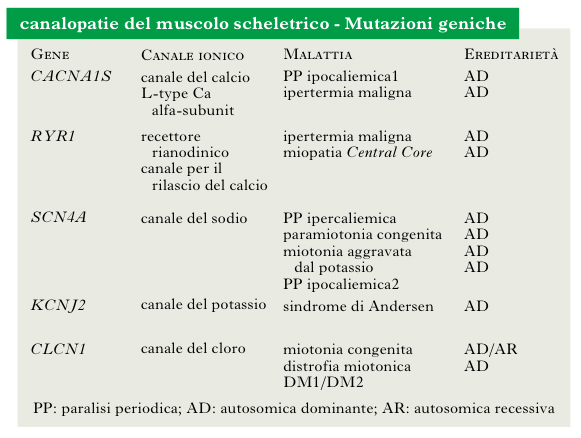
canalopatie del muscolo scheletrico
canalopatie del muscolo scheletrico
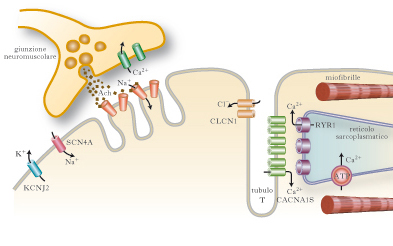
Malattie muscolari che derivano da alterazioni dei canali ionici situati a livello del sarcolemma e delle membrane interne della fibrocellula, e che intervengono nella generazione del potenziale d’azione, nel mantenimento della polarità della membrana e nell’avvio di processi endocellulari. Le mutazioni dei geni che codificano i canali ionici provocano miopatie ereditarie non destruenti (➔ miopatie), che presentano fra loro analogie cliniche e che sono state raggruppate nell’entità nosologica delle 80 canalopatie. Le canalopatie sono caratterizzate clinicamente da paralisi periodiche (per quanto riguarda le canalopatie del calcio o del sodio), ritardato rilasciamento dopo contrazione (➔ miotonia) e ipertermia maligna (canalopatie del calcio). Caratteristicamente, le differenti forme di paralisi periodica hanno una differente risposta alla somministrazione di potassio e di glucosio.
Canalopatie da alterazione del canale del sodio
L’alterata funzione dei canali per il sodio può determinare differenti quadri clinici. La paralisi periodica iperpotassiemica è una miopatia autosomica dominante dovuta a una mutazione del gene SCN4A che codifica per una proteina del canale del sodio. La paralisi compare in seguito alla somministrazione di potassio (o con il riposo dopo un esercizio fisico) e migliora con l’assunzione di glucosio. È dovuta a una ritardata inattivazione del canale del sodio, che prolunga l’entrata dello ione nella cellula dopo la depolarizzazione della membrana, provocando una persistente iperpolarizzazione e perdita di eccitabilità del sarcolemma. Ne consegue che il paziente può avere sia la miotonia, sia la paralisi. Il trattamento consiste nella somministrazione di carboidrati, salbutamolo, acetazolamide. La paralisi periodica iperpotassiemica può essere secondaria a insufficienza renale. La paramiotonia congenita è una forma di miotonia, autosomica dominante, che, contrariamente a quanto avviene nelle altre forme di miotonia, compare durante l’esercizio ripetuto. Nella paramiotonia il freddo determina caratteristicamente un peggioramento del fenomeno miotonico e la paralisi. Viene trattata con mexiletina. La miotonia aggravata dal potassio è una forma più lieve di miotonia, non sensibile al freddo, associata a diversi tipi di mutazione puntiforme del gene che codifica proteine per il canale del sodio.
Canalopatie da alterazione del canale del calcio
La paralisi periodica ipopotassiemica è una miopatia autosomica dominante, con un’incidenza di mutazioni de novo in un terzo dei casi. È caratterizzata da attacchi di paralisi muscolare generalizzata della durata di ore o giorni, in genere scatenati dal riposo dopo l’esercizio fisico, accompagnati da ipopotassiemia. La paralisi muscolare può essere molto marcata, fino alla completa tetraplegia, ma i muscoli respiratori sono risparmiati. Non si associa miotonia. È provocata nel 70% dei casi da una mutazione del gene CACNA1S, che codifica per la subunità alfa1 del canale del calcio. La penetranza è del 100% nei maschi e del 50% nelle femmine. Non è noto il meccanismo patogenetico che provoca la paralisi. L’ipertermia maligna è una rara ma grave e potenzialmente fatale condizione, che interviene in soggetti con un’anomalia genetica del recettore per la rianodina (RYR1), quando questi siano esposti, durante l’anestesia, a particolari farmaci scatenanti. Questi ultimi comprendono i vapori alogenati (fluotano, etrano, isofluorano) e i miorilassanti depolarizzanti (succinilcolina), ma teoricamente la complicanza può verificarsi con molti altri farmaci nei soggetti predisposti. L’affezione si manifesta con un quadro clinico caratterizzato da sintomi e segni che coinvolgono i muscoli (contratture e aumento della creatinfosfochinasi e della potassiemia), il sistema cardiocircolatorio (instabilità pressoria e alterazioni del ritmo cardiaco), la respirazione (dispnea, ipossia e ipercapnia), alterazioni del metabolismo (aumento ingravescente della temperatura corporea, da cui il nome di ipertermia), acidosi. La crisi viene trattata con infusione di dantrolene.
Canalopatie da alterazione dei canali del cloro
La miotonia congenita di Thomsen e la miotonia congenita di Becker (➔ miotonia) sono recessive e più comuni. Entrambe sono dovute a mutazione del gene CLCN1 sul cromosoma 7q35.10. La conduttanza del canale del cloro, che e dotato di due pori ionofori attraverso i quali passano gli ioni cloro, regola il potenziale di membrana a riposo. La mutazione di CLCN1 provoca una parziale depolarizzazione della membrana a riposo. Il quadro clinico e dominato dalla miotonia, dalla rigidità, dal warm-up (➔ miopatie) e dall’ipertrofia muscolare. La debolezza, quando presente, e lieve e prossimale. I pazienti presentano solitamente un notevole sviluppo delle masse muscolari.