
Biomateriali
Biomateriali
La definizione di biomateriale sulla quale attualmente esiste il più ampio consenso è quella stabilita nel corso della II International consensus conference on biomaterials, tenutasi a Chester (Inghilterra) nel 1991: «Si definisce biomateriale un materiale concepito per interfacciarsi con i sistemi biologici al fine di valutare, dare supporto o sostituire un qualsiasi tessuto, organo o funzione del corpo». Stret-tamente collegata a essa, occorre ricordare la definizione del cosiddetto dispositivo medico, contenuta nella Direttiva 93/42/CEE: «S’intende per dispositivo medico qualsiasi strumento, apparecchio, impianto, sostanza, o altro prodotto usato da solo o in combinazione, compreso il software informatico impiegato per il corretto funzionamento, e destinato dal fabbricante a essere impiegato nell’uomo a scopo di: diagnosi, prevenzione, controllo, terapia o attenuazione di una malattia; diagnosi, controllo, terapia, attenuazione o compensazione di una ferita o di un handicap; studio, sostituzione o modifica dell’anatomia o di un processo fisiologico; intervento sul concepimento, purché non eserciti l’azione principale nel o sul corpo umano, cui è destinato, con mezzi farmacologici o immunologici, né mediante processo metabolico, ma la cui funzione possa essere coadiuvata da tali mezzi».
La definizione di biomateriale sopra riportata si presta a diverse interpretazioni. L’interpretazione più restrittiva limita il concetto di biomateriale ai soli materiali strutturali. Bisogna però osservare che nel-la comunità scientifica mondiale un’interpretazione così limitata non è generalmente accettata. Un’interpretazione più ampia include frai biomateriali anche i materiali,soprattutto polimerici, usati come componenti di sistemi per il rilascio controllato di farmaci. Una terza interpretazione, ancora più ampia, estende il concetto di biomateriale anche ai polimeri solubili, che possono essere utilizzati come supporto molecolare di sostanze biologicamente attive. In effetti, attualmente uno dei settori di ricerca più avanzati è proprio quello che riguarda queste sostanze; si ricordino a titolo d’esempio alcune ricerche di straordinario interesse nel campo della cura dei tumori, nelle quali i polimeri solubili vengono impiegati per veicolare farmaci verso cellule o gruppi di cellule bersaglio, masse tumorali incluse.
La seconda e la terza interpretazione sopra ricordate relative al concetto di biomateriale in realtà coincidono. Infatti, in base alla seconda interpretazione, rientrano certamente in questa categoria oggetti impiantabili costituiti da materiali polimerici (lamine, cilindretti, aghi); tali oggetti contengono principî attivi che vengono rilasciati nel tempo e possono anche assumere la forma di microparticelle iniettabili. Con qualche modifica nella struttura dei polimeri usati, si possono anche preparare sistemi per il rilascio controllato di farmaci a base di particelle polimeriche di diametro inferiore a un micron (nanoparticelle), che non sarebbe giustificato escludere dalla categoria di biomateriale soltanto in base alle minori dimensioni. Ma anche addotti polimerici solubili di farmaci vengono studiati come sistemi particolarmente raffinati per il rilascio controllato di farmaci e pertanto, secondo la stessa logica, devono essere inclusi nella categoria di biomateriale. Questo è in effetti l’atteggiamento prevalentemente riscontrato nelle maggiori riviste internazionali di settore.
Definizioni
Biocompatibilità
L’attuale disponibilità di materiali, in termini di prodotti sia commerciali sia di ricerca, è talmente ampia da rispondere ai requisiti chimico-fisici e meccanici di quasi ogni applicazione biomedica concepibile. Tuttavia, sono pochi i materiali entrati in uso come biomateriali. Il principale fattore limitante è la biocompatibilità, perché un biomateriale è destinato per definizione a entrare in contatto con tessuti viventi.
La biocompatibilità può essere definita come la capacità di un materiale di non provocare da parte del sistema vivente nel quale è impiegato un insieme di reazioni sfavorevoli tali da pregiudicare la possibilità di utilizzarlo per tutto il tempo previsto. Un caso particolare di biocompatibilità è la emocompatibilità, cioè la compatibilità col sangue. Un requisito essenziale di un materiale emocompatibile è la sua non-trombogenicità, ossia esso non deve indurre la formazione di trombi quando viene in contatto col sangue (non deve farlo coagulare).
La biocompatibilità di un certo materiale deve essere valutata in base al tipo di prestazione richiesto. In particolare, occorre considerare la forma fisica, la sede dell’impianto e la sua durata, la presenza di additivi (come quelli plastificanti o stabilizzanti, nel caso di polimeri) e la natura degli eventuali prodotti di degradazione. Nel caso di un impianto medico, la biocompatibilità dipende evidentemente dallo stabilirsi di interazioni favorevoli fra i materiali che lo compongono, nella forma fisica che questi assumono, e i tessuti circostanti. L’esistenza di interazioni è prevista dalla definizione stessa di biomateriale; d’altronde, materiali che non suscitano reazioni di alcun genere da parte dell’organismo ospite sono adatti per applicazioni temporanee (per es., certi tipi di catetere) ma non per applicazioni a lungo termine (per es., protesi cardiovascolari od ortopediche). Infatti, affinché un impianto assicuri prestazioni stabili nel lungo periodo non basta che sia tollerato passivamente dall’organismo, ma deve essere in qualche modo incorporato, agganciato ai tessuti circostanti e comportarsi alla fine come un settore funzionale dell’organismo ospite. Questo presuppone che l’impianto subisca una colonizzazione cellulare e che le cellule colonizzanti vi aderiscano adeguatamente. Nei casi più favorevoli può accadere che l’impianto sia gradualmente ‘invaso’ dalle cellule e contemporaneamente degradi. Si forma così un tessuto autologo al posto del materiale originale del quale vengono mantenute la morfologia e la funzionalità.
La biocompatibilità di un materiale impiantato è influenzata, oltre che dalla sua tossicità, da numerosi fattori fra cui la forma dell’impianto, l’abilità del chirurgo che inserisce l’impianto, la dinamica dei moti dell’impianto una volta collocato, la biostabilità, cioè la resistenza alla degradazione chimica e meccanica, e la natura delle reazioni che avvengono all’interfaccia con i tessuti biologici. Questi fattori variano in modo significativo in funzione della collocazione dell’impianto (per es., se in tessuti duri o nel sistema cardiovascolare), tanto che la biocompatibilità può essere univocamente definita solo in funzione del tipo di applicazione. Inoltre, valutando la biocompatibilità di un materiale polimerico, bisogna evitare l’errore di confondere la sua tossicità intrinseca, cioè quella propria del polimero − spesso modesta o irrilevante − con quella di eventuali additivi che sono rilasciati nei tessuti circostanti dopo l’impianto.
Degradazione dei materiali in ambiente biologico
I tessuti e l’ambiente svolgono sui biomateriali diverse azioni. Qualsiasi alterazione chimica del materiale dovuta all’ambiente circostante che ne modifichi in modo sensibile le proprietà è detta degradazione, mentre si parla di biodegradazione se il materiale viene progressivamente degradato da attività biologiche.
La cosiddetta erosione consiste nella graduale dissoluzione di un materiale originalmente insolubile, do-vuta di solito a processi di degradazione non necessariamente mediati da attività biologiche specifiche; nel caso in cui tali azioni biologiche intervengano, è più appropriato parlare di bioerosione. In certi contesti, per indicare la scomparsa nel tempo del dispositivo dal sito d’impianto, si parla anche di bioassorbimento (riassorbimento, bioriassorbimento).
La bioeliminazione consiste nell’eliminazione del materiale dall’organismo ospite, generalmente dovuta a una degradazione che arriva fino al livello di piccole molecole, le quali sono espulse attraverso i normali processi d’escrezione.
Spesso si fa un uso non rigoroso dei termini biodegradazione e bioerosione, usati per indicare tutti i fenomeni di degradazione ed erosione che avvengono in ambienti biologici; tali termini presuppongono invece azioni biologiche specifiche, come l’intervento di enzimi, e si riferiscono a fenomeni che si verificano sia in ambienti biologici, sia in ambienti dove possono operare fattori biologici, per esempio il terreno. Se invece ad agire sono solamente fattori chimici e chimico-fisici − come la combinazione di temperatura e pH relativamente elevati con la presenza di acqua, sali e ossigeno − che, pur essendo tipici degli ambienti biologici, agirebbero allo stesso modo in qualunque altro ambiente, i termini corretti sono degradazione ed erosione. Negli impianti biodegradabili, i due tipi di processo possono avvenire contemporaneamente.
La bioeliminazione presuppone che il materiale sia, con il passare del tempo, completamente eliminato dall’organismo ospite. Ciò non avviene necessariamente in seguito alla dissoluzione del materiale dovuta a processi degradativi; esistono infatti materiali bioerodibili, ma non bioeliminabili: per esempio, il poli(2-idrossietilmetacrilato) ad alto peso molecolare (PHEMA) col tempo si trasforma in acido polimetacrilico, e quest’ultimo, se ad alto peso molecolare, è solubile, ma non eliminabile per escrezione renale.
La degradabilità di un materiale in ambiente fisiologico è una caratteristica che può essere sfavorevole o necessaria, ma sempre decisiva per la sua utilizzazione. Nel caso di dispositivi cui si richiedono prestazioni meccaniche di buon livello senza limiti di tempo (per es., protesi ortopediche, dentarie o cardiovascolari), il materiale ideale dovrebbe essere indegradabile e mantenere indefinitamente le sue proprietà. In altri casi è invece auspicabile, o addirittura obbligatorio, che il materiale sia non solo degradabile, ma anche bioeliminabile. Si possono citare come esempi i fili di sutura riassorbibili in chirurgia, fabbricati con speciali poliesteri, che non presuppongono operazioni di rimozione successive all’intervento; i polimeri bioerodibili usati come matrici per il rilascio controllato di farmaci; i materiali inorganici, polimerici o compositi allo studio come impalcature (scaffolds) per la rigenerazione tissutale; i polimeri solubili ad alto peso molecolare per veicolare sostanze biologicamente attive. Quando si prevede una degradazione del materiale dopo l’impianto del dispositivo, deve essere considerata l’eventuale attività biologica dei prodotti di degradazione. Occorre, infatti, che tali prodotti non siano sensibilmente tossici né in sede locale né in sede generale (in caso contrario il materiale non sarebbe biocompatibile) ed è preferibile che addirittura facciano normalmente parte dei costituenti dell’organismo o entrino nei normali cicli metabolici.
È infine opportuno rilevare che in campo biomedico non sono ammessi prodotti polimerici solubili, per quanto atossici, se non sono anche bioeliminabili. I polimeri solubili non sono escreti attraverso i reni se il loro peso molecolare supera una certa soglia, generalmente dell’ordine di qualche decina di migliaia. Quindi anche per l’uso di polimeri solubili il cui peso molecolare sia al di sopra di questa soglia è richiesta la biodegradabilità. La non completa bioeliminabilità ha determinato l’abbandono come sostituto del plasma del poli-N-vinilpirrolidinone (polivinilpirrolidone o povidone), polimero praticamente non degradabile in ambiente fisiologico, nonostante tutte le altre proprietà, biocompatibilità inclusa, fossero estremamente favorevoli a tale scopo.
2. Classificazione dei biomateriali
Classificazione in base alla provenienza e alla struttura chimica
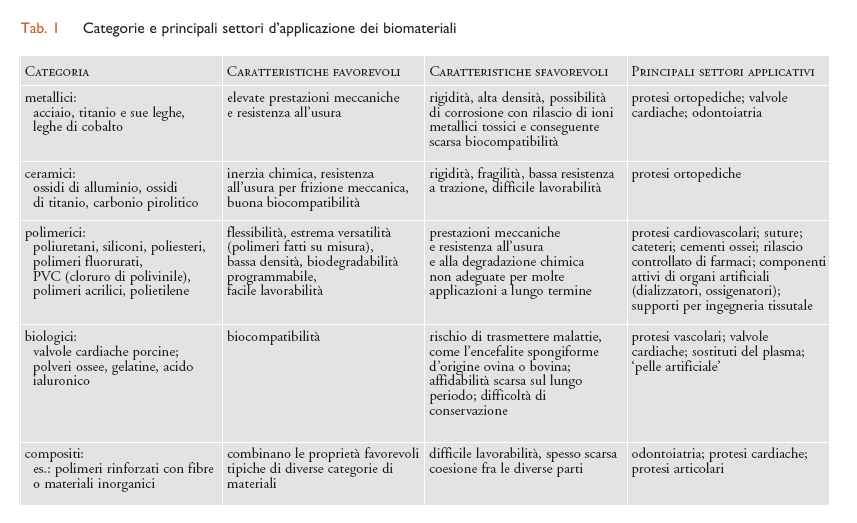
I biomateriali possono essere classificati in modo del tutto analogo ai materiali in genere. Possiamo quindi distinguere cinque categorie di biomateriali: metallici, ceramici, polimerici, biologici e compositi, intendendo per questi ultimi associazioni fisiche fra materiali diversi, per esempio polimeri rinforzati con cariche inorganiche. Ognuna di queste categorie presenta caratteristiche proprie, che ne rendono conveniente l’uso in applicazioni mediche specifiche (tab. I).
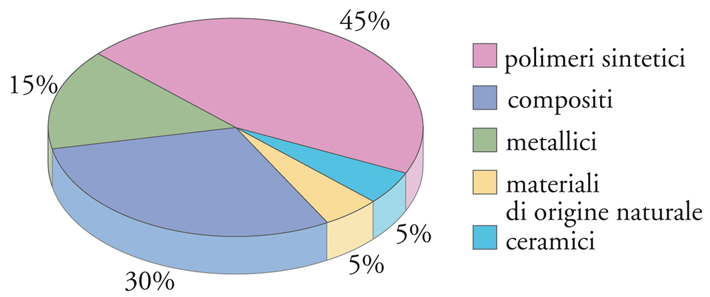
La proporzione d’uso fra le diverse categorie di biomateriali, relativamente ai prodotti attualmente sul mercato (escludendo cioè i prodotti di ricerca), è mostrata in fig. 2. Si può osservare che i biomateriali polimerici rappresentano da soli quasi la metà del totale e, considerando che, dal punto di vista della struttura chimica, i biomateriali d’origine biologica sono quasi tutti di tipo polimerico, questa percentuale è in realtà ancora maggiore. A ciò contribuiscono vari fattori, fra i quali l’estrema versatilità dei polimeri relativamente alle loro caratteristiche fisico-chimiche, l’ampiezza delle conoscenze accumulate negli ultimi decenni riguardo al rapporto fra struttura e proprietà, e infine la relativa facilità di sintesi in laboratorio. Ciò permette di programmare strutture polimeriche su misura per la risoluzione di determinati problemi medici, prepararle e sperimentarle su scala ridotta e, in caso di successo, trasferirne la produzione su scala industriale. Tuttavia, i materiali inorganici, metallici e ceramici, restano insostituibili per tutte le applicazioni che necessitano di elevate prestazioni meccaniche, soprattutto in termini di durezza superficiale, rigidità e resistenza all’usura.
Classificazione in base al tipo di applicazione
A seconda del tipo di applicazioni cui sono destinati, i biomateriali possono essere classificati in quattro categorie principali che saranno esaminate separatamente: (a) biomateriali strutturali; (b) materiali bioerodibili che servono come impalcature per la rigenerazione di tessuti; (c) componenti di sistemi per il rilascio controllato di farmaci; e (d) veicolanti solubili di sostanze biologicamente attive.
Inoltre, rientrano nel concetto di biomateriale anche i materiali, per lo più polimerici, che compongono attrezzature medico-chirurgiche: quali, per esempio, tubi, fili di sutura, cateteri per applicazioni temporanee, contenitori per sangue.
Biomateriali strutturali
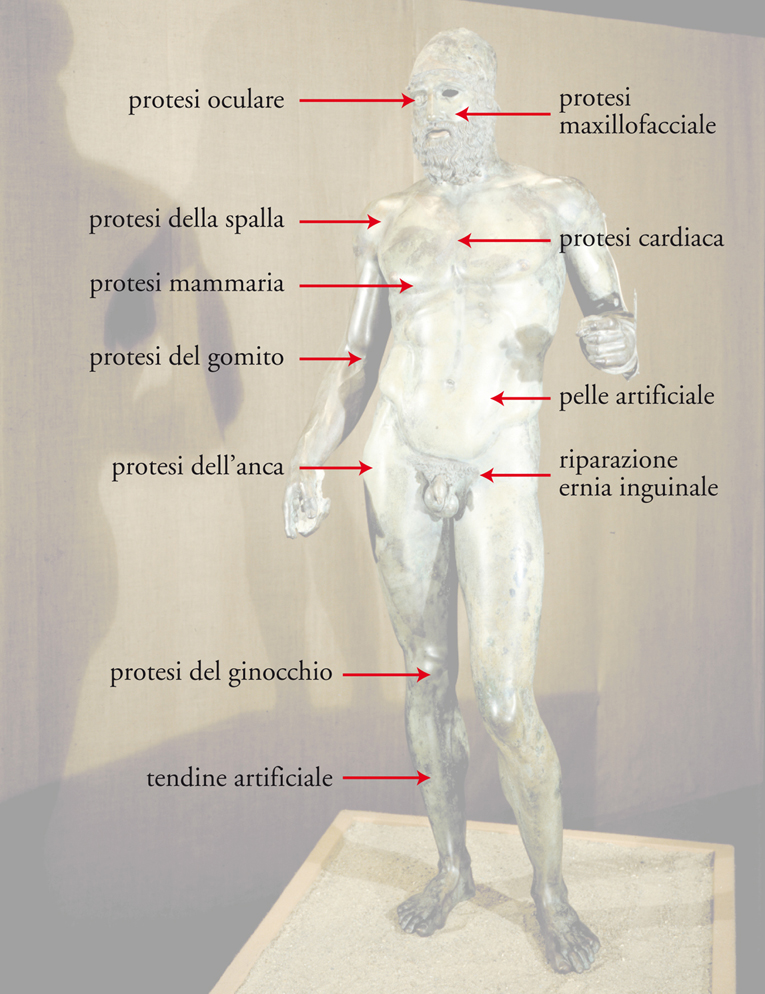
I biomateriali strutturali sono destinati a riparare o sostituire tessuti, o anche interi organi del corpo umano, la cui funzionalità è compromessa da incidenti, malattie, usura dovuta all’età, o in qualche caso malformazioni genetiche. Devono essere inclusi in questa categoria anche i sostituti sintetici del plasma e del sangue, perché questi in campo medico sono considerati tessuti a tutti gli effetti. Il numero e la complessità degli interventi di sostituzione attualmente possibili possono apparire stupefacenti. Un’indicazione in proposito, seppure non esauriente, è data dalla fig. 3.
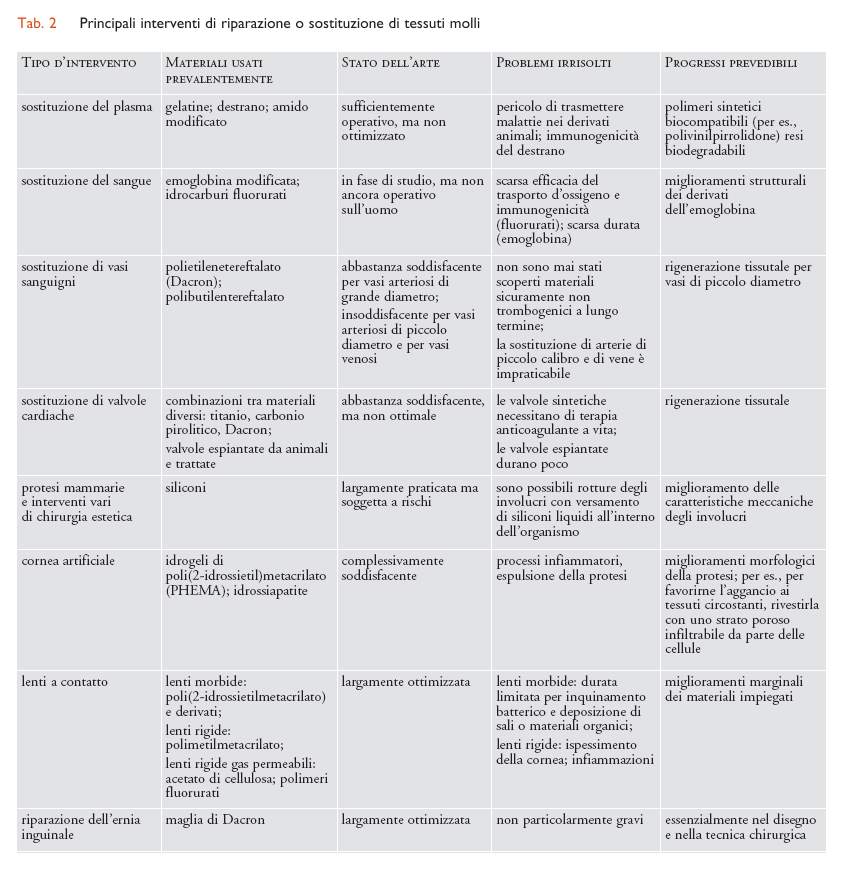
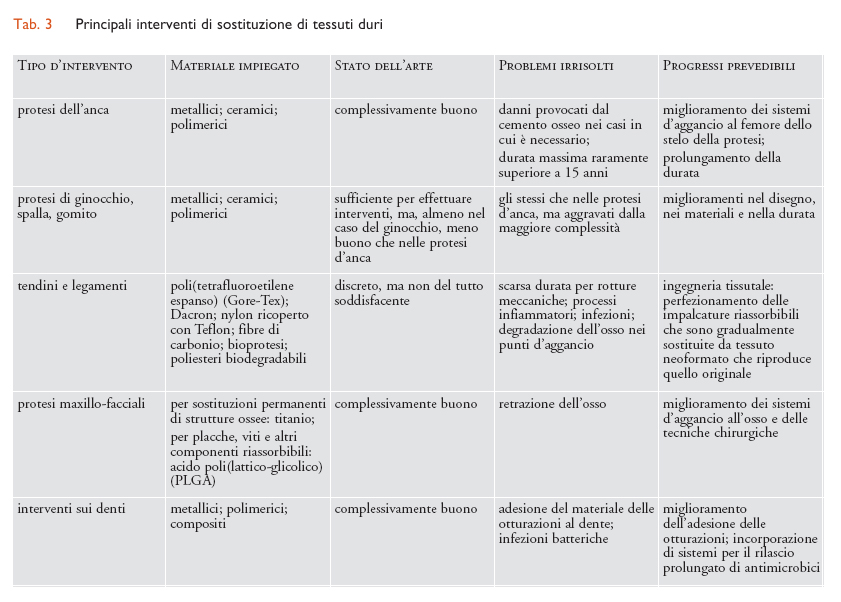
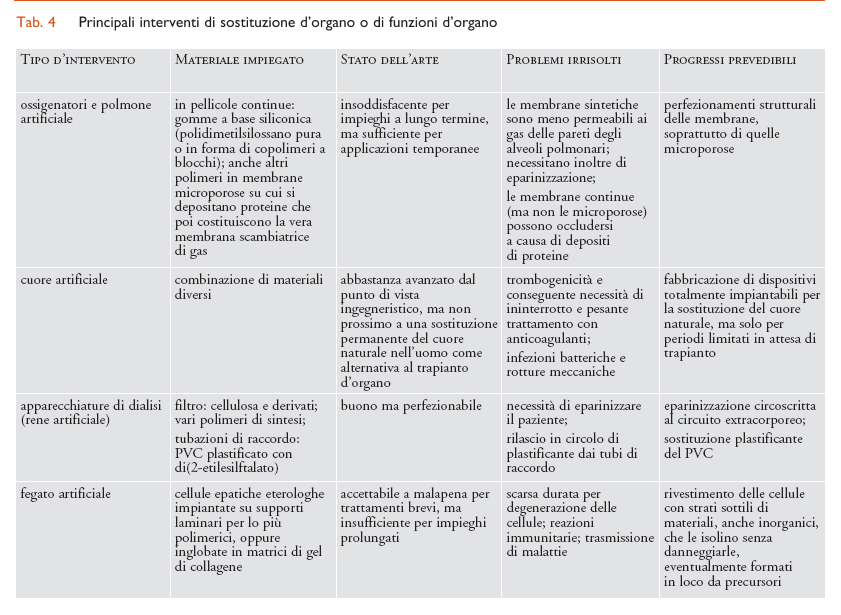
Considerando il tipo di tessuto interessato, i molteplici interventi di sostituzione che impiegano biomateriali strutturali possono essere suddivisi a grandi linee in tre settori: tessuti molli (plasma e sangue inclusi), tessuti duri e organi complessi, ognuno dei quali richiede l’uso di diverse categorie di biomateriali. In particolare, per la sostituzione di tessuti molli sono prevalentemente impiegati biomateriali polimerici; per quella di tessuti duri sono per lo più utilizzati materiali metallici o ceramici; per organi complessi sono usati tutti i tipi di materiali, in proporzioni diverse secondo l’organo interessato. Dove si richiedono funzioni non esclusivamente meccaniche (per es., permeabilità selettiva a gas o soluti) il componente attivo è per lo più di natura polimerica. I principali interventi di riparazione o sostituzione per ciascuno dei tre settori sopra indicati sono riassunti nelle tabb. 2, 3 e 4, insieme con i biomateriali impiegati caso per caso.
Per dare qualche esempio rilevante delle problematiche connesse con l’uso di biomateriali strutturali sono stati scelti per una trattazione più estesa i seguenti tipi d’intervento, considerati di particolare interesse sociale: (a) le protesi d’anca; (b) i sostituti sintetici del plasma e del sangue; (c) le protesi cardiovascolari; (d) il cuore artificiale; (e) il rene artificiale.
Protesi d’anca
Gli impianti ortopedici costituiscono sicuramente uno dei principali settori di applicazione dei biomateriali per quanto riguarda il numero degli interventi e il fatturato annuo, che solo nel nostro Paese è dell’ordine di 200 milioni di euro. Esiste una grande varietà d’impianti ortopedici. Accanto agli impianti protesici per i quali tecniche e materiali sono ormai ampiamente consolidati – per esempio le protesi d’anca, di ginocchio, di spalla e di altre articolazioni – si possono ricordare i cosiddetti mezzi di sintesi per applicazioni temporanee, quali placche, viti, fissatori esterni e altri di uso corrente nella pratica clinica, e infine i tendini e le cartilagini artificiali, tuttora oggetto di una notevole attività di ricerca i cui risultati, soprattutto per le seconde, non sono ancora ottimali.
Fra i molteplici impianti ortopedici in uso, il caso della protesi totale d’anca è sicuramente tra i più diffusi e di maggiore impatto sociale. L’articolazione dell’anca, detta anche articolazione coxo-femorale, funziona come un giunto sferico formato dalla testa del femore e dalla coppa (acetabolo) che la ospita. I movimenti rotatori ammissibili hanno angoli limitati sia dalla presenza di legamenti e muscoli, sia dalla struttura a labbro dell’acetabolo, che assicura stabilità all’accoppiamento.
Una protesi d’anca ideale deve rispondere ai seguenti requisiti: (a) deve adempiere alla sua funzione principale, vale a dire consentire gli stessi movimenti dell’articolazione naturale, e sopportare gli sforzi relativi; (b) deve resistere alla fatica meccanica e all’usura per un tempo superiore all’attesa di vita del paziente (si noti che l’articolazione dell’anca, in un soggetto che svolge un’attività normale, è sottoposta a circa un milione di cicli per anno); (c) deve essere biocompatibile; (d) deve poter essere sottoposta a microlavorazioni che ne consentano la migliore adattabilità al sito d’impianto.
Le moderne protesi d’anca riproducono con notevole fedeltà la struttura dell’articolazione naturale. Esse sono tipicamente composte da quattro elementi: (a) uno stelo metallico; (b) una testa, o cotile, in materiale metallico o ceramico, che sostituisce l’originale testa del femore; (c) una coppa acetabolare in materiale polimerico; (d) un guscio acetabolare metallico che funge da mezzo d’unione rigido fra la coppa acetabolare e la struttura ossea del bacino.
Stelo. - Lo stelo è attualmente fabbricato con classi ben definite di leghe metalliche; speciali materiali compositi attualmente allo studio sono ancora lontani dall’essere impiegati nell’uomo. Le leghe metalliche attualmente applicate sono a base di cobalto e titanio; fra queste ultime si può citare la lega Ti-6Al-4V. Leghe in cui il vanadio è sostituito dal niobio o dal ferro, considerate più biocompatibili, hanno un uso più limitato. Le leghe metalliche, e in particolare le leghe di titanio, presentano l’inconveniente di essere soggette a corrosione per sfregamento, con conseguente metallosi (effetti tossici dovuti all’immissione in circolo di ioni metallici) e necessitano di adeguati trattamenti superficiali di anodizzazione.
Normalmente lo stelo è inserito nel canale diafisario del femore, cui è in qualche modo ancorato. Per ottenere una corretta distribuzione delle forze, un buon ancoraggio, ottenibile tramite una corretta progettazione dello stelo stesso e la scelta di un conveniente sistema d’ancoraggio, deve di preferenza essere localizzato nella zona prossimale dello stelo. A seconda del tipo di ancoraggio, le protesi si distinguono in cementate e non cementate.
Nelle protesi cementate s’interpone fra lo stelo e le pareti del canale diafisario una miscela di metilmetacrilato monomero e di polimetilmetacrilato, che viene aggiunto per ridurre il ritiro dovuto alla polimerizzazione e per rendere viscosa la miscela, con una certa percentuale di cariche inorganiche inerti (zirconia o solfato di bario). Per mezzo di un sistema catalitico redox, generalmente costituito da un perossido e un’ammina aromatica terziaria (la dimetiltoluidina), si induce poi la polimerizzazione in loco del monomero. Alla fine del processo la massa del cemento, cariche inorganiche a parte, è costituita interamente da polimetilmetacrilato e lo stelo è agganciato alle pareti dell’osso. Si impiegano di solito da 30 a 40 grammi di cemento per protesi.
Tutti gli elementi della miscela, tranne il polimero preformato, sono tossici, in particolare i perossidi e l’ammina. La polimerizzazione sviluppa calore e, nonostante questo sia smaltito in parte dall’organismo, si raggiungono facilmente nei tessuti immediatamente circostanti temperature superiori a 60 °C, con conseguenti fenomeni necrotici. Questo sistema d’ancoraggio è rimasto sostanzialmente immutato fin dalle prime applicazioni, e stupisce che in oltre trent’anni non ne sia stato introdotto uno migliore.
Inizialmente le protesi cementate erano la maggioranza e, malgrado i loro inconvenienti, rappresentano ancora circa un terzo del totale. Esse sono impiegate soprattutto in soggetti anziani o in interventi ripetuti, perché in questi casi la rigenerazione dell’osso attorno allo stelo è più difficile.
Nelle protesi non cementate si tenta di realizzare un accoppiamento preciso fra stelo e canale diafisario, e si sfrutta la capacità dell’osso di rigenerarsi e ancorarsi direttamente alla superficie metallica. L’ancoraggio è favorito dall’impiego di steli porosi, o da alcuni trattamenti superficiali. Si può rivestire lo stelo, per esempio, con microsfere o filamenti metallici porosi, oppure ricoprirlo con idrossiapatite, che è gradualmente riassorbita e sostituita da tessuto osseo di nuova formazione.
Testa protesica. - In passato testa protesica e stelo costituivano un pezzo unico, ma recentemente si preferisce costruirli separatamente e unirli in un secondo tempo mediante accoppiamento conico. Questa tecnica ha il vantaggio di poter realizzare i due componenti in materiali diversi e di adattarne meglio le rispettive dimensioni alla struttura fisica del paziente.
Le teste possono essere di tipo metallico o ceramico. Le teste metalliche sono per lo più in lega Ti-6Al-4V, in lega Co-29Cr-6Mo, o in acciaio inossidabile ASTM F138. Sono più economiche di quelle ceramiche, ma vannosoggette a corrosione e usura. Le teste ceramiche sono realizzate in allumina o zirconia stabilizzata. Nonostante una certa fragilità e un maggior costo rispetto alle teste metalliche, presentano importanti vantaggi, in quanto non vanno soggette a corrosione e sono meglio bagnabili dai fluidi organici lubrificanti prodotti dall’organismo (soluzioni di acido ialuronico), assicurando così un minore coefficiente d’attrito e di conseguenza una minore usura della coppa acetabolare.
Coppa e guscio acetabolare. - Le coppe sono attualmente realizzate in polietilene ad altissimo peso molecolare (Ultra high molecular weight polyethylene, UHMWPE), che rispetto ad altri possibili materiali resiste particolarmente bene all’usura. L’UHMWPE ha totalmente sostituito per quest’uso il Teflon che, a causa delle sue scarse caratteristiche meccaniche, si usurava rapidamente, con formazione di detriti, nonostante l’inerzia chimica. Anche nel caso di coppe acetabolari in UHMWPE, però, l’usura non è trascurabile e rappresenta forse il problema più difficile da risolvere se si vuole portare la durata delle protesi a oltre 20 anni, mentre attualmente è di circa 15 anni. Per ridurre l’usura della coppa acetabolare si può ricorrere a trattamenti superficiali di finitura della testa femorale se questa è metallica, ma soprattutto sostituire le teste femorali metalliche, più soggette a usura, con teste ceramiche.
La coppa acetabolare è connessa con l’osso del bacino tramite un guscio acetabolare metallico che viene serrato su di essa in modo da formare un tutto unico. Generalmente il guscio non è cementato, anche se abbinato a uno stelo cementato, e le tecniche per assicurarne la connessione con l’osso sono le stesse descritte nel caso degli steli non cementati.
Sostituzione di plasma e sangue
I sostituti sintetici del plasma e del sangue sono preparati farmacologici in grado di vicariarne le principali funzioni e vengono utilizzati in caso di ipovolemia sanguigna. L’organismo umano, infatti, ha la capacità di compensare piccole perdite di sangue mediante rigenerazione spontanea. Se però il volume totale del sangue scende al di sotto di un certo valore limite, come avviene per esempio in seguito a ustione o grave trauma, può insorgere nell’organismo una sindrome definita shock emorragico, vale a dire un complesso di reazioni dovute a uno squilibrio emodinamico acuto che coinvolge tutti i processi metabolici negli organi vitali.
Lo shock emorragico può essere limitato intervenendo con una trasfusione sanguigna. Nella pratica, però, la trasfusione omologa di sangue presenta un gran numero di problemi, causati, per esempio, dalla poca disponibilità di sangue, dal fatto che il sangue ha scarsa stabilità (può essere conservato a 4 °C per un massimo di tre settimane) e dai rischi biologici legati alla possibilità di infezioni virali (in particolare AIDS ed epatiti), di sensibilizzazione antigenica e di incompatibilità di gruppo sanguigno. Questi motivi pratici, uniti alla diffusione di convinzioni religiose che vietano la trasfusione, spiegano la necessità di sostituti sintetici del plasma o del sangue.
Sostituti sintetici del plasma (plasma expanders). - Il plasma costituisce la fase liquida del sangue e ne rappresenta circa il 55% in volume. Esso è costituito per il 92% da acqua e per il 7% da proteine (albumine, fibrinogeno e globuline), vari ioni e sostanze organiche (urea, acido urico, lipidi, amminoacidi). I sostituti del plasma sono solitamente soluzioni colloidali polimeriche che presentano proprietà, come pressione osmotica e viscosità, molto simili a quelle del plasma. I prodotti più largamente utilizzati sono il destrano (un polisaccaride di origine microbica), le gelatine (prodotti di origine proteica ottenuti mediante idrolisi parziale di collagene animale) e l’amido modificato. Ciascuno di questi prodotti è però caratterizzato da specifici effetti collaterali. Le gelatine, utilizzate in più del 70% dei casi, comportano una serie di rischi biologici connessi alla loro origine animale; il destrano, oltre a essere leggermente antigenico, interferisce, se somministrato in grandi quantità, con la coagulazione e le funzioni renali. Si può ricordare che, storicamente, il primo polimero usato in soluzione per la sostituzione del plasma è stato il poli-N-vinilpirrolidone, o PVP, ad alto peso molecolare, introdotto attorno al 1940 e adottato su larga scala dagli eserciti combattenti, salvando centinaia di migliaia di vite senza inconvenienti postumi dimostrabili. Questo polimero avrebbe proprietà complessivamente superiori rispetto a tutti gli altri sostituti del plasma attualmente in uso, ma non è interamente bioeliminabile ed è stato per questo motivo abbandonato.
Sostituti sintetici del sangue. - Il sangue è costituito, oltre che dal plasma, da una parte corpuscolata, formata essenzialmente da globuli bianchi, globuli rossi (deputati al trasporto d’ossigeno che effettuano tramite una proteina, l’emoglobina) e piastrine. Formulazioni in grado di veicolare ossigeno sono solitamente definite sangue artificiale. Tali preparati, anche se non riescono a espletare la maggior parte delle attività immunologiche e coagulative del sangue, rappresentano un’alternativa all’infusione di sangue o di frazioni ematiche. I principali trasportatori d’ossigeno finora studiati come sostituti del sangue possono essere classificati in due categorie: le emulsioni di perfluorocarburi e le soluzioni di emoglobina modificata.
Emulsioni acquose di perfluorocarburi (PFC) a basso peso molecolare mostrano un’elevata capacità di disciogliere ossigeno. Il contenuto in ossigeno, e quindi la capacità di trasporto, delle emulsioni di PFC dipende dalla pressione parziale dell’ossigeno (PO2) e gli studi finora effettuati hanno dimostrato che per raggiungere tensioni fisiologiche d’ossigeno sono solitamente necessarie atmosfere con elevato contenuto d’ossigeno, talvolta fino al 90÷100%. Nel 1968 un’emulsione di perfluorotributilammina (C12F27N) venne utilizzata per sostituire completamente il sangue di ratti, i quali sopravvissero in atmosfera di ossigeno quasi puro. Tuttavia, la tendenza dei perfluorocarburi a essere catturati dalle cellule del sistema reticoloendoteliale ha limitato lo sviluppo clinico di questi sostituti del sangue.
Soluzioni diluite di emoglobina pura mostrano un’elevata capacità di trasporto d’ossigeno, ma sono filtrate rapidamente attraverso i glomeruli renali, e quindi escrete, come conseguenza della dissociazione dell’emoglobina in prodotti di peso molecolare inferiore. Inoltre, l’elevata affinità dell’emoglobina per l’ossigeno ne rende poco efficace il rilascio nei tessuti dai vasi capillari. La polimerizzazione dell’emoglobina mediante glutaraldeide ha condotto a un notevole incremento del suo tempo di ritenzione nel sistema vascolare. La difficoltà a rilasciare ossigeno ai tessuti viene attualmente ridotta combinando l’emoglobina con 2,3-difosfoglicerato, che ne limita la tendenza a ritenere l’ossigeno.
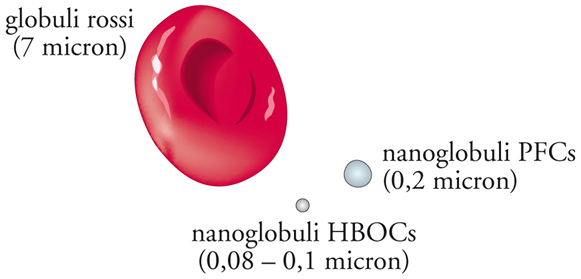
Le compagnie farmaceutiche hanno sviluppato alcuni tipi di sangue artificiale a partire dal 1980, ma la maggior parte di loro ha abbandonato le ricerche in questo campo dopo che in esperimenti sull’uomo si erano verificati attacchi cardiaci, ictus e morti. Alcune formulazioni dei primi tempi causavano anche collasso delle pareti capillari ed eccessivo innalzamento della pressione sanguigna. Tuttavia, ricerche successive hanno prodotto diversi sostituti del sangue appartenenti a due classi: trasportatori di ossigeno a base emoglobinica (HBOC) e perfluoroidrocarburi (PFC) (fig. 5).
Alcuni fra questi trasportatori di ossigeno sono prossimi alla fine della fase di studio e presto potrebbero essere disponibili negli ospedali. Altri sono già in uso. Un HBOC chiamato commercialmente Hemopure è, per esempio, correntemente usato negli ospedali sudafricani dove la diffusione dell’AIDS rende problematica la disponibilità di sangue per le trasfusioni. Un trasportatore di ossigeno basato su PFC chiamato Oxygent è alle ultime fasi di sperimentazione sull’uomo in Europa e in America settentrionale.
La struttura chimica dei due tipi di trasportatori d’ossigeno è molto diversa, ma entrambi funzionano sopprattutto per diffusione passiva dell’ossigeno. La diffusione passiva dipende dalla tendenza dei gas a passare dalle aree di maggior concentrazione a quelle a concentrazione minore fino a raggiungere uno stato di equilibrio. Le soluzioni di HBOC ricordano il sangue. Sono rosso scure e sono costituite da vera emoglobina sterilizzata ottenuta da fonti diverse, come globuli rossi di sangue umano che ha oltrepassato i limiti di scadenza, placente umane, globuli rossi bovini o batteri geneticamente modificati in grado di produrla.
Tuttavia, non è possibile semplicemente iniettare l’emoglobina nel circolo del paziente. L’emoglobina è molto efficiente nel trasportare e rilasciare l’ossigeno quando è protetta dalle membrane cellulari, ma se è libera degrada rapidamente, e i suoi prodotti di degradazione causano gravi danni renali. Per questo motivo, gli HBOC impiegano forme modificate di emoglobina, più resistenti alla degradazione della molecola naturale. Alcune delle tecniche più comuni usate a questo scopo sono: stabilire legami fra parti diverse della molecola dell’emoglobina (reticolazione), la polimerizzazione dell’emoglobina legando fra di loro svariate molecole; preparare coniugati fra l’emoglobina e polimeri sintetici. Sono stati anche descritti sistemi in cui l’emoglobina è racchiusa in una membrana sintetica a base di lipidi, colesterolo o acidi grassi. Un HOBC, denominato MP4, è costituito da emoglobina rivestita da catene di polietilenglicol (PEG).
Protesi vascolari
La sostituzione di segmenti di grossi vasi arteriosi con protesi artificiali rappresenta uno dei più antichi e tuttora riusciti impieghi di materiali sintetici in campo medico-chirurgico. Si racconta che poco dopo il 1960 il chirurgo statunitense Michael E. DeBakey, disperando di salvare un paziente colpito da aneurisma aortico, si strappasse un lembo di camicia, casualmente di polietilenetereftalato (Dacron), lo cucisse a forma di tubo e lo innestasse sostituendo il tratto d’arteria compromesso. Il paziente si salvò. Il polietilenetereftalato è tuttora un materiale molto usato per questo tipo di protesi.
Le protesi vascolari odierne si dividono, per quanto riguarda l’origine, in due gruppi principali: protesi biologiche e protesi sintetiche. Le protesi biologiche derivano dalla sostituzione dei tratti danneggiati con vasi d’analogo diametro ricavati da organismi viventi, opportunamente trattati. Esse possono essere ulteriormente suddivise in protesi autologhe od omologhe (vena safena, vena ombelicale, arteria mammaria, arteria iliaca) e protesi eterologhe (carotide bovina). Le protesi sintetiche sono ricavate da polimeri di sintesi. Si suddividono a loro volta, secondo la lavorazione, in tessili e non tessili: queste ultime sono costituite principalmente da politetrafluoroetilene espanso (Gore-Tex, ePTFE), hanno discreta emocompatibilità e possono essere usate per vasi di diametro relativamente modesto, ma sono piuttosto deboli dal punto di vista meccanico.
Sembra opportuno descrivere in maggior dettaglio le protesi tessili, in quanto sono di gran lunga le più usate. Esse sono generalmente costituite da polietilenetereftalato, a volte trattato superficialmente con carbonio pirolitico per aumentarne la biocompatibilità. Possono essere distinte in tre tipi: (a) protesi a rete, che presentano una trama a graticcio con le fibre perpendicolari fra loro; (b) protesi a maglia, che presentano una trama ad anelli intrecciati; (c) protesi a velour, caratterizzate da una trama fibrillare solo esterna (monovelour), oppure sia interna sia esterna (doppio velour).
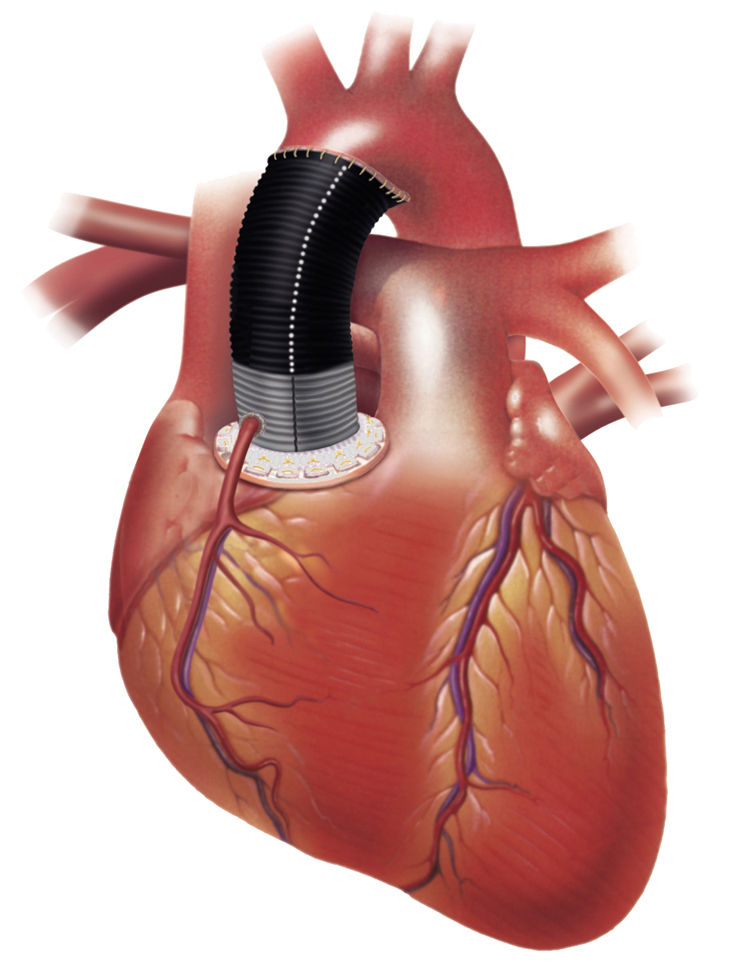
Le protesi tessili prima dell’uso sono sottoposte a tre processi: compattazione, pieghettatura e pulitura. La compattazione, consistente nell’applicazione di calore o di solventi, conduce a un restringimento della trama con diminuzione della porosità e stabilizzazione della struttura. La pieghettatura permette alle protesi di seguire i movimenti del corpo piegandosi senza strozzarsi – consentendone l’impiego anche in corrispondenza di parti mobili, come per esempio le articolazioni – e le rende estensibili, quindi operativamente più facili da impiantare. Inoltre, poiché tali protesi sono dotate di elasticità longitudinale, riescono ad adattarsi alle pulsazioni del flusso sanguigno, diminuendo di conseguenza le sollecitazioni nei punti di sutura con l’arteria naturale (fig. 6). La pulitura è fatta meccanicamente o con solventi. In quest’ultimo caso occorre che siano completamente eliminati dalla superficie del vaso gli eventuali residui di solvente. La sterilizzazione delle protesi di Dacron, necessaria come per tutte le protesi, viene generalmente effettuata con ossido di etilene o raggi gamma.
Le protesi tessili sono più o meno porose a seconda del tipo, e quelle a rete sono le più compatte. Sono tutte fortemente trombogeniche, e ciò compensa per certi aspetti gli inconvenienti della porosità, dato che dopo l’impianto il sangue coagula occludendo gli interstizi fra le maglie e assicurando così una rapida emostasi. Per assicurare la tenuta delle protesi a maglia e a velour, che sono più porose, occorre effettuare un processo di pre-coagulazione in modo da occludere preventivamente i pori e non avere importanti perdite ematiche iniziali.
Nei casi favorevoli, dopo l’impianto la protesi viene ricoperta da un tessuto che simula le pareti dei vasi normali (neointima), la cui vitalità dipende dalla vascolarizzazione delle protesi, che è più difficile nel caso di quelle meno porose. Inoltre, se il vaso artificiale è poco poroso, la neointima può tendere a distaccarsi. D’altro lato, se il vaso artificiale è molto poroso, è difficile assicurarne la tenuta iniziale. Per superare questo dilemma sono entrate in commercio protesi molto porose, le cui pareti non sono inizialmente permeabili essendo impregnate con polimeri biodegradabili, di solito d’origine naturale (per es., collagene, albumina o derivati dell’elastina), destinati dopo l’impianto a essere gradualmente sostituiti dal tessuto di neoformazione. Per accelerare la formazione di quest’ultimo, un metodo ancora in fase di studio utilizza cellule endoteliali ottenute dalle vene o dalle cellule mesoteliali del peritoneo, che sono inseminate sulla matrice trombotica.
Le principali complicazioni osservate dopo l’impianto dipendono dal tipo di protesi. Le protesi biologiche invecchiano rapidamente e possono mineralizzarsi, sclerotizzarsi lungo le pareti, degradare, o assorbire lipidi. Le protesi tessili possono infettarsi, disinserirsi, o, soprattutto il tipo a doppio velour, degradare. Possono inoltre andare incontro a cedimenti meccanici, quali dilatazione o rottura. Con relativa frequenza, a causa di un’incompleta formazione della neointima, o del distacco di questa, oppure di fenomeni di necrosi, possono formarsi trombi anche successivamente allo stadio iniziale.
Infine, è evidente da quanto sopra esposto che i vasi arteriosi di grande diametro possono essere sostituiti con protesi artificiali, perché in queste ultime la formazione dello strato trombotico si arresta prima che il lume interno si occluda. Vasi arteriosi di diametro inferiore ai 6 mm o vasi venosi (quindi con flusso sanguigno relativamente lento e maggiore facilità di formazione di trombi) non possono invece essere sostituiti, perché allo stato attuale non esistono biomateriali sufficientemente emocompatibili e le eventuali protesi col tempo si occluderebbero.
In tempi recenti si è verificata un’accelerazione nello sviluppo di nuovi materiali e nuove tecniche chirurgiche a minima invasività, iniziando un’era terapeutica per combattere le malattie cardiovascolari caratterizzata dal posizionamento di stent, e l’uso di laser e rotoablatori per rimuovere ostruzioni del lume dei vasi sanguigni. L’inserzione di stent coronarici rappresenta attualmente l’80÷90% di tutti gli interventi alle coronarie, coinvolgendo milioni di pazienti di tutto il mondo.
I materiali più usati attualmente per costruire gli stent sono metalli. Il più utilizzato, per esempio, è l’acciaio inossidabile, indubbiamente molto funzionale nel mantenere immediatamente pervio il vaso ripristinando il flusso sanguigno. Tuttavia, gli stent metallici hanno serie limitazioni, come indurre la formazione di trombi, e talvolta non adattarsi per dimensioni al vaso. Inoltre, sono permanenti. Dopo la re-endotelizzazione della parete interna del vaso diventano inutili e anzi possono provocare infiammazioni croniche o ripresentare a lungo termine il problema della formazione di trombi anche se questa era stata inizialmente tenuta sotto controllo. Queste limitazioni hanno indotto a progettare stent costituiti da materiali biodegradabili.
Materiali polimerici a memoria di forma rappresentano una categoria con elevate potenzialità applicative. Questi materiali, se subiscono un cambiamento di forma in seguito a una sollecitazione esterna, hanno la caratteristica di ricordare la forma primitiva e di poterla ancora riassumere. Il caso più generale è costituito da materiali che possono assumere due diverse forme interconvertibili. Esistono però materiali ‘intelligenti’ che possono assumere tre forme diverse, come per esempio un copolimero a segmenti fra poli-ε-caprolattone (PCL) e poli-cicloesilmetacrilato (PCHMA) chiamato MACL. Questa capacità di assumere a seconda delle condizioni tre forme diverse rende possibili innumerevoli applicazioni tecnologiche, in particolare in campo medico. Uno stent costituito, per esempio, da un materiale ‘intelligente’ può essere introdotto in un vaso sanguigno in una forma compatta e poi espandersi nel sito di impianto fino ad assumere una forma funzionale per il dispositivo medico. In caso di necessità, la sua rimozione sarà facilitata riducendone le dimensioni fino a renderlo facilmente maneggevole, per esempio tramite sollecitazioni termiche.
Valvole cardiache
La sostituzione delle valvole cardiache è un tipo d’intervento ormai effettuato normalmente e con buone prospettive di successo. La valvola aortica, posta fra il ventricolo sinistro del cuore e l’aorta, è la più esposta ad alterazioni dovute a diverse cause e, di conseguenza, è la più frequentemente sostituita con protesi.
La valvola cardiaca ideale dovrebbe avere una serie di caratteristiche. Dovrebbe essere facilmente posizionata, adattarsi ai movimenti naturali del cuore, non essere immunogenica, permettere un flusso regolare del sangue senza causare danni ai suoi costituenti, durare indefinitamente o almeno molto a lungo, non essere rumorosa per non peggiorare la qualità della vita del paziente. Nessuna fra le valvole impiegate attualmente soddisfa tutti questi requisiti. Di conseguenza, non esiste la valvola cardiaca ideale per tutti i pazienti, ma occorre scegliere per ciascun paziente il tipo di valvola più adatto alle sue esigenze e al suo stato.
Le valvole attualmente impiegate possono essere divise in due gruppi: biologiche (o bioprotesi) e meccaniche.
Le protesi biologiche sono costituite da tre lembi valvolari di tessuto biologico opportunamente trattato, generalmente d’origine suina, sostenuto nei tipi più tradizionali da un’armatura di materiali sintetici rivestita di polietilentereftalato (protesi stented). Più recentemente sono state introdotte protesi biologiche che non hanno l’armatura di materiale sintetico (protesi stentless), le quali hanno dimensioni ridotte e sono applicabili anche a persone di corporatura minuta. Le protesi valvolari biologiche hanno vantaggi e svantaggi analoghi a quelli delle corrispondenti protesi vascolari. Il principale vantaggio è costituito dalla loro bassa trombogenicità, che in molti casi permette di evitare la terapia anticoagulante, rendendole particolarmente indicate per pazienti che presentano controindicazioni per questo tipo di terapia. Il loro principale svantaggio sta nel fatto che esse hanno scarsa durata, in quanto il tessuto biologico può andare incontro a degenerazione, e ciò le rende poco adatte per pazienti con una lunga prospettiva di vita.
I tipi di protesi meccaniche attualmente più usati sono costituiti da tre elementi: un anello d’inserzione ricoperto di teflon o polietilentereftalato, cui è attaccata una struttura metallica contenente un elemento mobile che può essere una palla, un disco o due emidischi; in quest’ultimo caso le protesi assicurano un flusso laminare centrale e mostrano le migliori caratteristiche emodinamiche. Struttura di sostegno ed elemento mobile sono a volte rivestiti di carbonio pirolitico per migliorarne l’emocompatibilità. Le protesi meccaniche hanno una durata generalmente superiore a quella delle protesi biologiche, e questo è il principale vantaggio che offrono; esse presentano però una elevata trombogenicità, e pertanto, benché la velocità del flusso sanguigno attraverso le protesi valvolari riduca l’entità del problema rispetto ad altri tipi di protesi, il paziente non può evitare di sottoporsi permanentemente a una terapia anticoagulante.
Cuore artificiale
Per cuore artificiale (TAH, Total artificial heart) si intende un cuore completamente artificiale che viene impiantato dopo rimozione totale o parziale del cuore. Un TAH deve necessariamente contenere i seguenti componenti: (a) un’unità di comando (interna e/o esterna), cioè un sistema di controllo elettronico della modalità di pompaggio, eventualmente programmabile dall’esterno e in grado di regolare la funzione di pompaggio in relazione all’attività del paziente; (b) una pompa, per fornire la necessaria spinta al sangue; (c) un convertitore d’energia, per convertire l’energia erogata dal particolare tipo d’alimentazione impiegato nell’energia spesa per il pompaggio; (d) un alimentatore, per fornire l’energia (elettrica, meccanica, nucleare) necessaria per il pompaggio del sangue. L’alimentatore è generalmente extracorporeo, talvolta portatile, e il trasferimento d’energia al dispositivo impiantato può essere di tipo pneumatico, meccanico, con fili elettrici, o con trasformatore. Dato l’elevato numero di componenti, vengono ovviamente impiegati biomateriali di diverse classi, in particolare polimeri, metalli, ceramiche e carbonio pirolitico.
Un cuore artificiale ideale dovrebbe avere alcune caratteristiche essenziali: anzitutto essere capace di fornire la portata ematica opportuna, adattandola in ogni momento alle esigenze fisiologiche dell’organismo; poi dovrebbe essere biocompatibile (in particolare non dovrebbe causare emolisi, né formazione di trombi, né dare origine a infezioni), avere un’elevata resistenza alla fatica e, infine, dovrebbe essere facile da impiantare, portatile e poco costoso.
Dal punto di vista meccanico, il cuore artificiale sarebbe già realizzabile in pratica. Gli aspetti non ancora ottimizzati sono la bassa durata delle batterie, non superiore a due anni, e la possibile rottura di alcuni componenti meccanici. Sono difficoltà in linea di principio superabili, perché non sembra impossibile predisporre sia fonti d’energia ricaricabili o rinnovabili, sia sistemi per il monitoraggio dell’esaurimento o dell’usura delle varie parti, sia interventi di sostituzione delle parti non più affidabili. Un problema più serio è invece quello dell’insufficiente biocompatibilità nel medio/lungo termine dei dispositivi finora realizzati o realizzabili nel prossimo futuro.
Il cuore artificiale Jarvik-7, il primo sperimentato sull’uomo nel 1982, era formato da due ventricoli separati con camere d’aria ed era azionato da un sistema pneumatico. I ventricoli del cuore presentavano una superficie liscia ed erano in poliuretano segmentato (Biomer®). I diaframmi erano costituiti da fogli di Biomer a quattro strati altamente flessibili. I due ventricoli avevano una forma sferica, e le aree di passaggio ai grandi vasi e agli atri erano poste in posizione anatomica. Le quattro valvole a disco che assicuravano un flusso di sangue unidirezionale erano in carbonio pirolitico. I manicotti atriali che provvedevano alla connessione con gli atri naturali erano realizzati in tessuto di Dacron. I manicotti erano uniti al cuore artificiale mediante sistemi di connessione costituiti da segmenti rivestiti da policarbonato rigido. Protesi vascolari di Dacron provvedevano alla connessione con i grandi vasi.
I materiali impiegati nel cuore artificiale totale Jarvik-7, in relazione alle caratteristiche fisiche necessarie alle loro funzioni, erano, e sono ancora, i più biocompatibili fra quelli noti. Nonostante ciò, nelle numerose sperimentazioni su animali la principale causa di morte nei primi 30 giorni d’impianto è stata l’emorragia, dovuta sicuramente alla terapia anticoagulante necessaria per evitare la formazione di trombi. Una seconda causa di morte erano le infezioni, mentre la morte per fallimento meccanico, in particolare delle valvole o del diaframma, è risultata largamente minoritaria.
La trombogenicità di un dispositivo è funzione sia dei biomateriali impiegati, sia delle loro caratteristiche di superficie (quest’ultima, a parità di altre condizioni, è tanto meno trombogenica quanto più è liscia), sia del disegno dell’apparecchiatura per quanto riguarda le caratteristiche di fluidodinamica.
Il cuore artificiale Utah-100, progettato a Salt Lake City, rappresenta un’evoluzione del Jarvik-7, il cui disegno è stato perfezionato per migliorarne l’affidabilità soprattutto relativamente alla formazione di trombi. Dai test su animali da esperimento è risultato un tempo medio di sopravvivenza di 78 giorni, con 14 animali sopravvissuti oltre 60 giorni e un massimo di sopravvivenza di 331 giorni. Le cause di morte sono state globalmente all’incirca le stesse che nel Jarvik-7, e il miglioramento per quanto riguarda la formazione di trombi è stato complessivamente modesto. Altri cuori artificiali, per esempio il modello AbioCor costruito dalla ditta Ambiomed in Massachusetts, sono attualmente in fase di sperimentazione sull’uomo.
La maggior parte dei gruppi di ricerca coinvolti attribuisce il fallimento a lungo termine di tutti i cuori artificiali finora impiantati alle infezioni e alla possibilità di guasti meccanici, mentre di solito la trombogenicità dei materiali non è messa in evidenza. Quest’ultima, invece, costituisce il principale fattore che limita nel tempo l’utilità pratica del cuore artificiale totalmente impiantabile, perché rende necessaria, dopo l’intervento, una continua e pesante terapia anticoagulante non tollerabile dal paziente (o dall’animale) per lunghi periodi.
Si può forse migliorare ulteriormente il disegno di un sistema così complesso, ma non eliminare il problema della trombogenicità dei materiali; e non sembra facile, dopo circa trent’anni di insuccessi, realizzare nuovi materiali intrinsecamente non trombogenici per un tempo indefinito. Ne consegue che la realizzazione di un cuore artificiale adatto a sostituire definitivamente l’organo naturale nell’uomo, vale a dire tale da costituire un’alternativa al trapianto d’organo, con ogni probabilità non è imminente. Sembra invece meno remota la realizzazione di un cuore artificiale capace di assicurare la sopravvivenza, per tempi relativamente lunghi, di pazienti privi della funzione cardiaca naturale e in attesa di trapianto. Sono d’altra parte già in uso dispositivi d’assistenza, per esempio dispositivi d’assistenza ventricolare (VAD), che possono servire sia come aiuto al recupero di una funzione cardiaca momentaneamente compromessa, sia come sistema temporaneo in attesa del trapianto in situazioni di scompenso cardiaco grave. Un VAD non va però inteso come sostituzione d’organo, perché viene messo in parallelo al cuore per aiutarlo, ma non lo sostituisce.
Rene artificiale
Le due soluzioni attualmente possibili per risolvere i casi di insufficienza renale sono il trapianto d’organo e l’emodialisi.
Mentre il trapianto presenta i tipici limiti dell’insufficiente numero dei donatori e della possibilità di rigetto, l’emodialisi rappresenta uno dei più grandi successi della medicina moderna, grazie al quale decine di migliaia di pazienti in Italia, e centinaia di migliaia nel mondo, vivono una vita quasi normale, mentre in altri tempi sarebbero stati condannati a sofferenze e morte precoce.
L’emodialisi è un processo per la purificazione del sangue in pazienti nei quali la funzione renale è compromessa. Il suo scopo è sostituire la funzione del rene, che consiste nell’eliminazione sia dell’acqua in eccesso, sia dei metaboliti (come l’urea) il cui accumulo darebbe origine a effetti tossici, sia di sostanze tossiche accidentalmente presenti. Questo scopo è raggiunto tramite lo scambio di soluti e acqua fra il sangue e una soluzione (liquido di dialisi), scambio che avviene attraverso una membrana polimerica semipermeabile (membrana dializzante o membrana ultrafiltrante) contenuta in un filtro dializzatore.
Un’apparecchiatura di dialisi (il cosiddetto rene artificiale) è una macchina piuttosto complessa, costituita da quattro strutture principali: il filtro dializzatore, il circuito ematico extracorporeo, il circuito del liquido di dialisi e il sistema di monitoraggio.
Il filtro dializzatore è l’unità funzionale dell’apparecchiatura; esso contiene una membrana semipermeabile che permette l’attraversamento, e quindi lo scambio con il liquido di dialisi, di sostanze al di sotto di una certa soglia di dimensioni molecolari, all’incirca la stessa consentita dalla filtrazione renale in soggetti sani, mentre le macromolecole proteiche restano nel circolo sanguigno. La velocità di scambio fra sangue e liquido di dialisi è proporzionale all’area della superficie della membrana e alla sua porosità, e questi parametri possono essere variati entro ampi limiti. Nei primi modelli la forma del filtro era a foglietti e, per assicurare un’adeguata superficie di scambio, era necessario costruire filtri molto voluminosi; i filtri più moderni sono invece costituiti da un fascio di fibre cave, le quali consentono di ottenere una grande superficie di scambio in una struttura compatta
Le membrane semipermeabili possono essere cellulosiche e sintetiche. Le membrane cellulosiche sono costituite da cellulosa rigenerata o da cellulose modificate per esterificazione, hanno buone caratteristiche generali e sono relativamente economiche, ma richiedono una più intensa terapia anticoagulante per evitare la formazione di trombi. Attualmente, le membrane cellulosiche vengono gradualmente sostituite da quelle sintetiche, costituite da polimeri sintetici diversi a seconda del tipo: polisulfoni, poliacrilonitrile modificato per aggiunta di co-monomeri anionici (per es., acido acrilico o alfa-metilallilsolfonico), polimetilmetacrilato, copolimeri etilene-alcol vinilico, policarbonato o poliammidi. Le loro prestazioni sono buone e riproducibili per quanto riguarda l’efficacia e la velocità di scambio, ed esse risultano in genere più emocompatibili delle membrane cellulosiche, ma sono meno economiche.
Recentemente sono state studiate e introdotte nell’uso altre tecniche di purificazione del sangue complementari o alternative all’emodialisi − l’emodiafiltrazione, la biofiltrazione e l’emofiltrazione − che utilizzano soprattutto le membrane sintetiche con maggiore capacità depurativa e permeabilità all’acqua.
L’emofiltrazione non utilizza liquido di scambio e applica al sangue essenzialmente lo stesso principio (osmosi inversa) usato in uno dei principali processi di desalinizzazione dell’acqua di mare. Il sangue che scorre attraverso il filtro è sottoposto a pressione, e la differenza di pressione fra sangue e ambiente esterno provoca il passaggio attraverso la membrana, e quindi l’eliminazione, di acqua e soluti a basso peso molecolare. Per mantenere costante il volume ematico del paziente, l’ultrafiltrato è continuamente sostituito da una soluzione elettrolitica bilanciata infusa sia nella linea d’ingresso sia in quella d’uscita del sangue dall’emofiltro.
Tanto l’emodialisi quanto le tecniche alternative necessitano che il paziente sia sottoposto a trattamento anticoagulante. L’anticoagulante generalmente usato per evitare la formazione di trombi all’interno del circuito di dialisi è l’eparina. La tendenza alla formazione di trombi, con la conseguente necessità di trattamento anticoagulante, costituisce nei dializzati cronici un importante fattore di mortalità per emorragie e infarti miocardici. Un’eparinizzazione limitata al solo circuito extracorporeo potrebbe evitare questi inconvenienti e potrebbe essere realizzata con resine biocompatibili in grado di riassorbire l’eparina prima che il sangue dal circuito extracorporeo venga reimmesso nell’organismo. Sistemi di questo tipo sono ancora in una fase sperimentale.
Nelle apparecchiature di dialisi, come in molte altre apparecchiature mediche, il circuito ematico extracorporeo è costituito da una serie di tubi in cloruro di polivinile (PVC) plastificato con di-(2-etilesil)ftalato, comunemente denominato DHEP o DOP. Questo materiale presenta importanti vantaggi pratici, quali l’economicità e la facilità di lavorazione e di assemblaggio, potendosi saldare ad altre parti dello stesso materiale in modo rapido e perfetto solo inumidendo con opportuni solventi le parti da saldare e accostandole, senza aggiunta di collanti. Inoltre, in confronto con le altre parti dell’apparecchiatura di dialisi, il PVC plastificato non è particolarmente trombogenico. Tuttavia, vi è un certo rilascio di DHEP nel sangue del paziente, e questa sostanza, un tempo considerata inerte, viene attualmente ritenuta responsabile di azioni biologiche negative, fra le quali tossicità a livello della divisione cellulare e riduzione della fertilità. Sono allo studio, ma non ancora d’uso corrente, altri materiali, fra cui PVC plastificato con additivi diversi, non facilmente estraibili e biocompatibili.
Oltre ai due tipi di intervento descritti, sono allo studio nuovi approcci nell’ambito dell’ingegneria tissutale, che sarà trattata nel paragrafo seguente. In particolare, si tratta della preparazione di tubuli bioartificiali, tramite la coltivazione di cellule epiteliali all’interno di fibre cave permeabili sia all’acqua sia alle sostanze disciolte. I primi esperimenti sono stati effettuati seminando cellule epiteliali renali di cane (MDCK) all’interno di un’unica fibra cava. Successivamente, si è passati a un reattore a fibre multiple, dove le cellule tubulari mantenevano non solo le proprietà di trasporto, ma anche varie funzioni endocrine e metaboliche, utilizzando cellule renali prelevate da maiale. Combinando questo bioreattore con una cartuccia sintetica per emofiltrazione nel circuito extracorporeo si sono ripristinate le funzioni di filtrazione, di trasporto ed endocrinologiche del rene in cani con uricemia acuta. Questo però non significa che l’applicazione clinica del dispositivo sia prossima. Anzitutto, occorrerebbe ripetere gli esperimenti usando cellule umane per non andare incontro a insormontabili problemi di rigetto; inoltre, le fibre cave rivestite internamente di cellule epiteliali hanno una grande tendenza a occludersi per la formazione di trombi. Fra i possibili materiali polimerici formabili in forma di membrane o fibre cave come supporti per l’adesione e proliferazione delle cellule epiteliali, le prime ricerche sembrano indicare che poliacrilonitrile e polisolfoni siano i più promettenti.
Ingegneria tissutale
Con l’espressione tissue engineering, ingegneria tissutale, si intende la ricrescita di tessuto vivente usando come impalcatura (o matrice) un biomateriale preferibilmente riassorbibile o asportabile dopo che la sua funzione è terminata. Dopo alcuni tentativi sporadici risalenti alla prima metà del Novecento, l’interesse per questa tecnica è ripreso verso il 1970 e ha avuto uno sviluppo che si può definire straordinario a partire dal 1990. Un esempio di ricerca avanzata basato su tecniche di ingegneria tissutale è stato anticipato nel paragrafo precedente.
L’ingegneria tissutale e il trapianto selettivo di cellule nacquero come mezzi per sostituire tessuti malati con tessuti viventi progettati e realizzati per soddisfare i requisiti individuali di ogni paziente. Lo scopo finale è ripristinare, mantenere o anche migliorare le funzioni di un determinato tessuto con l’impianto di elementi viventi che diventano parte integrante del paziente stesso.
Il metodo più comune di ingegneria tissutale consiste nell’uso di combinazioni cellule-matrice in sistemi che possono essere aperti o chiusi. Un sistema aperto inizia con la coltivazione in vitro di cellule isolate, preferibilmente appartenenti allo stesso paziente. Le cellule sono poi inseminate sopra un’impalcatura naturale o sintetica dove continuano a proliferare e dopo un sufficiente intervallo di tempo il sistema viene impiantato nel paziente. Crescita, proliferazione e differenziamento successivi delle cellule possono essere controllati mediante la scelta del tipo di materiale e l’aggiunta di opportuni fattori. Nel caso di cellule provenienti dallo stesso paziente, il vantaggio di questa tecnica consiste nell’evitare i problemi di biocompatibilità a lungo termine, spesso riscontrati invece con l’innesto di materiali sintetici; a processo ultimato, infatti, il tessuto originario è rimpiazzato da uno virtualmente identico. Un sistema chiuso differisce per il fatto che le cellule sono isolate dal corpo da una membrana semipermeabile che permette lo scambio dei nutrienti e l’eliminazione dei rifiuti metabolici, ma protegge le cellule da azioni avverse, incluse eventuali risposte immunitarie.
L’ingegneria tissutale dà di norma risultati migliori rispetto a due possibili tecniche alternative più semplici, anch’esse allo studio e in alcuni casi applicate. La prima consiste nell’iniettare una sospensione di cellule contenente fattori di crescita direttamente sulla lesione, senza uso d’impalcature; tuttavia, la mancanza di immobilizzazione può consentire alle cellule di migrare dal sito d’impianto. La seconda tecnica consiste nell’impiantare l’impalcatura lasciando che le cellule già presenti nell’organismo la infiltrino, analogamente a quanto accade nella saldatura spontanea all’osso del femore degli steli porosi delle protesi d’anca non cementate. Questa tecnica, però, esponendo la superficie del materiale dell’impalcatura direttamente ai tessuti, può provocare reazioni da corpo estraneo, che sono invece evitate se si applica l’impalcatura preventivamente colonizzata da cellule provenienti dallo stesso organismo ospite.
L’impalcatura ideale dovrebbe essere biodegradabile, in modo che dopo la guarigione rimanga solo tessuto nativo, ma questo richiede che la velocità di biodegradazione corrisponda a quella della rigenerazione del tessuto. Essa dovrebbe inoltre possedere proprietà chimiche ideali per promuovere l’adesione cellulare e l’angiogenesi, nonché per controllare il differenziamento cellulare in modo che il tessuto di neoformazione sia identico a quello originale. Ciò può essere realizzato aggiungendo alterreno di coltura adeguati fattori di crescita. Infine, vi dovrebbe essere un elevato rapporto fra superficie e volume, in modo da permettere un’alta densità di colonizzazione da parte delle cellule.
Le impalcature comunemente impiegate nell’ingegneria tissutale possono avere varie forme, quali tessuti e fibre, impianti porosi con pori chiusi o aperti, membrane. Le impalcature fibrose biodegradabili sono generalmente costituite da materiali polimerici a carattere di poliesteri, come l’acido poliglicolico o copolimeri fra acido lattico e acido glicolico (PLA). Sono però usate anche fibre di materiali non biodegradabili, come per esempio fibre di carbonio. Le impalcature porose, usate soprattutto in ortopedia e in chirurgia ricostruttiva, possono impiegare, oltre agli stessi polimeri biodegradabili, anche materiali inorganici riassorbibili, quali fosfati di calcio o biovetri. Impalcature a forma di membrana, utilizzate per esempio per la rigenerazione del derma, sono spesso formate da polimeri di origine naturale, quali collagene o acido ialuronico, opportunamente modificati in modo da renderli insolubili in acqua, ma fortemente idrofilici e capaci, se posti in un mezzo acquoso, di formare pellicole e membrane rigonfiabili e morbide.
Rilascio controllato di farmaci
Il rilascio controllato di farmaci è una tecnica che permette di liberare nell’organismo sostanze biologicamente attive con velocità prevedibile e terapeuticamente congrua. L’obiettivo generalmente perseguito è una cinetica di rilascio di ordine zero, in altre parole una velocità di rilascio costante nel tempo fino a quando la carica di principio attivo non si è esaurita. Se invece ci si limita a prolungare nel tempo il rilascio di una sostanza attiva, con scarso controllo sulla cinetica, è preferibile usare l’espressione rilascio sostenuto, o anche, come talvolta viene chiamato, rilascio prolungato, rilascio nel tempo, rilascio programmato.
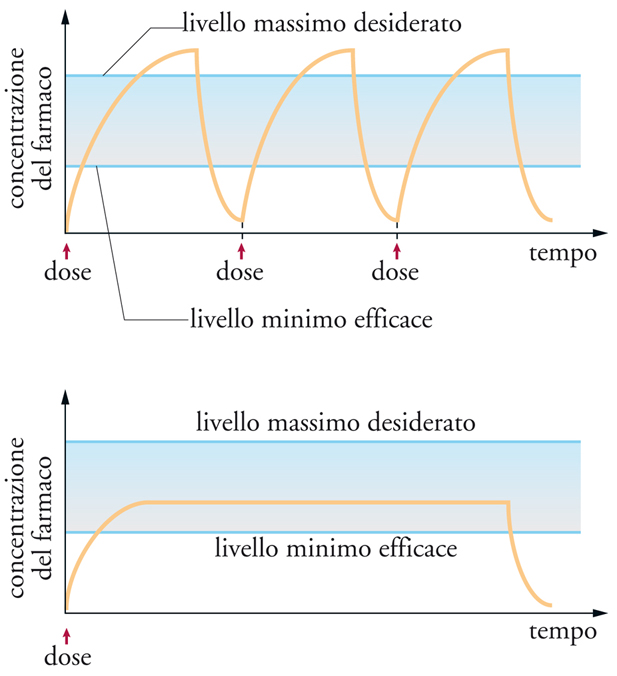
Ogni farmaco, in linea di principio, ha un determinato intervallo di concentrazione entro il quale è terapeuticamente utile. Concentrazioni più alte o più basse risultano, rispettivamente, in effetti tossici o inattività. L’ampiezza dell’intervallo di concentrazione utile (la cosidetta finestra terapeutica) varia entro ampi limiti ed è tipica per ogni farmaco. Anche se qualsiasi farmaco col tempo viene completamente eliminato, la sua scomparsa per escrezione o per trasformazione chimica − in altre parole, la sua permanenza nell’organismo − avviene con una velocità tipica per ciascun farmaco. Ormoni peptidici o peptido-mimetici sono per esempio eliminati tanto rapidamente da essere in pratica inutili sul piano terapeutico se somministrati con metodi tradizionali, e pertanto vengono sempre più diffusamente prescritti in abbinamento con opportuni sistemi di rilascio controllato. Lo scopo di questa tecnica, infatti, è quello di evitare le somministrazioni ripetute di farmaci con le conseguenti oscillazioni di concentrazione, e di mantenere quest’ultima all’interno della finestra terapeutica per lunghi periodi. Una rappresentazione grafica della differenza fra i due tipi di somministrazione è riportata in fig.12.
I sistemi per il rilascio controllato di farmaci (drug delivery systems) sono costituiti essenzialmente da materiali polimerici e possono essere suddivisi in due categorie: dispositivi di varia complessità non bioeliminabili, che liberano il farmaco per diffusione o con altri meccanismi fisici, e dispositivi semplici bioeliminabili, dai quali il farmaco è rilasciato sia per diffusione, sia per progressiva erosione del dispositivo. I dispositivi della prima categoria, una volta esauriti, debbono essere rimossi.
Dispositivi basati sulla diffusione
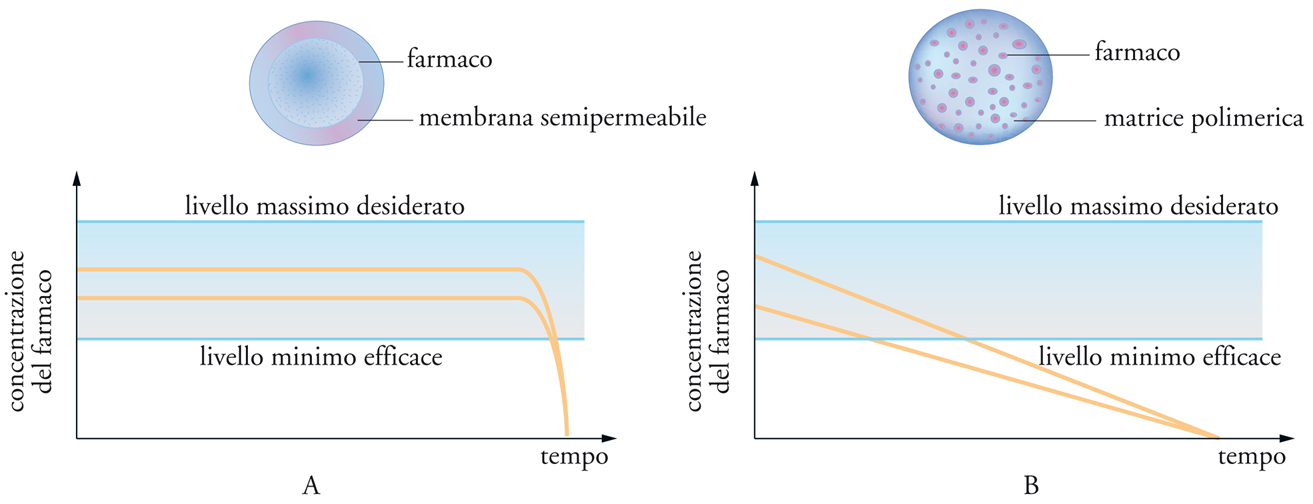
I sistemi per il rilascio controllato basati sulla diffusione si possono dividere in sistemi a serbatoio (reservoir devices), in cui il farmaco è incapsulato entro una membrana inerte, e sistemi monolitici (monolithic devices), in cui il farmaco è disperso o sciolto in una massa di polimero inerte. Dettagli sul loro funzionamento sono mostrati nella fig. 13. Si può osservare che nei primi è almeno teoricamente possibile realizzare una velocità di rilascio di ordine zero, indipendente dalla quantità residua di farmaco nel dispositivo, mentre nei secondi è più corretto parlare di rilascio sostenuto. Esistono attualmente numerosi tipi commerciali di dispositivi a serbatoio, che sono usati in modo particolare per la somministrazione di ormoni o di farmaci per uso oftalmico.
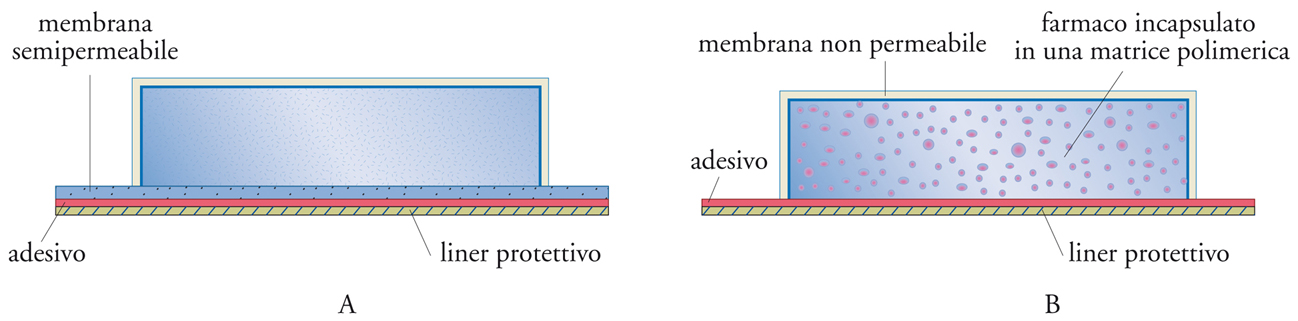
Alla categoria dei sistemi a rilascio per diffusione appartengono anche i dispositivi per la somministrazione transdermica (cerotti), che possono essere monolitici o a serbatoio. Si tratta di dispositivi relativamente complessi, schematicamente rappresentati in fig. 14. È importante che in questi dispositivi lo stadio lento sia l’attraversamento della membrana, e non della pelle, per assicura-re un rilascio del farmaco prevedibile e indipendente sia dalle caratteristiche individuali del paziente, sia dal suo stato fisico.
Rilascio controllato da fenomeni di degradazione
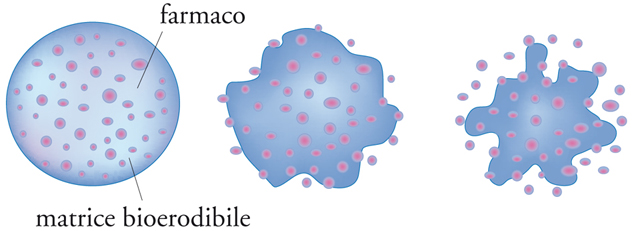
Nei sistemi a rilascio controllato dalla degradazione, il farmaco è disperso in una matrice polimerica degradabile e riassorbibile che può assumere forme diverse, quali lamine o cilindretti impiantabili, aghi, microsfere iniettabili. Il polimero viene gradualmente eroso, perché contiene nella catena principale dei legami sensibili alla degradazione idrolitica, rilasciando il farmaco nell’ambiente circostante. Alla fine del processo tutto il materiale scompare (fig. 15).
È opportuno osservare che in realtà una parte del farmaco viene rilasciato, soprattutto all’inizio, anche attraverso un meccanismo di diffusione che si sovrappone alla degradazione della matrice. I polimeri degradabili maggiormente usati come matrici appartengono alla classe dei poliesteri carbossilici; altri polimeri attualmente allo studio come matrici degradabili sono, per esempio, le polianidridi, i poliacetali, i poliortoesteri, il chitosano e i suoi derivati, i polifosfazeni.
La degradazione idrolitica consiste nella scissione di legami covalenti per azione dell’acqua, di solito con l’assistenza di altre specie chimiche presenti in soluzione. La degradazione idrolitica dei poliesteri a contatto con il sangue o altri liquidi biologici può essere mediata da enzimi, o semplicemente dovuta alle caratteristiche dell’ambiente acquoso in cui si vengono a trovare. Nel caso dei materiali polimerici di uso più comune (acido polilattico, acido poliglicolico o loro copolimeri), entrambi i meccanismi hanno luogo contemporaneamente. Da un certo punto di vista, è preferibile che la degradazione dei materiali sia causata prevalentemente dall’ambiente, le cui caratteristiche, a differenza delle attività biologiche, variano poco sia da individuo a individuo, sia in rapporto alle condizioni fisiche di ogni individuo. Ciò consente di predeterminare con precisione la velocità di degradazione dopo l’impianto, la quale dipende soltanto dalla forma e dalla struttura chimica dei materiali. Si possono stabilire correlazioni fra struttura e velocità di degradazione; infatti, a parità di gruppo chimico idrolizzabile, tale velocità diminuisce all’aumentare della cristallinità e delle caratteristiche idrofobiche, e ciò spiega perché i poliesteri sopra ricordati, relativamente idrofilici, siano degradabili e bioeliminabili, mentre il polietilenetereftalato (Dacron), anch’esso appartenente alla classe dei poliesteri, ma di struttura fortemente idrofobica, sia invece stabile in ambiente fisiologico. È per tale ragione che esso costituisce il materiale più usato per la fabbricazione di protesi vascolari.
Il processo di rilascio del farmaco e di riassorbimento del dispositivo può durare, secondo i casi, da pochi giorni a molti mesi, con risultati in un certo senso spettacolari: ormoni peptidici e peptido-mimetici quali LHRH o suoi analoghi di sintesi (essenziali nella cura del tumore della prostata), che iniettati tal quali avrebbero una vita media nell’organismo misurabile in minuti, se inseriti in matrici bioerodibili forniscono in certi casi concentrazioni costanti e terapeuticamente congrue per un anno e oltre.
Nanoparticelle
Recentemente sono stati introdotti sistemi di rilascio controllato basati su particelle di polimeri bioerodibili (nanoparticelle) di dimensioni tipicamente comprese fra 0,1 e 0,5 µm, caricate con il principio attivo, le quali hanno una migliore penetrazione e circolazione all’interno del corpo rispetto a particelle più grandi e possono es-sere somministrate senza problemi attraverso iniezioni endovenose o intramuscolari. Le nanoparticelle, infatti, anche se insolubili in solventi acquosi vi rimangono di regola perfettamente sospese, e a occhio nudo le lorosospensioni hanno l’aspetto di soluzioni.
Un ulteriore vantaggio di questa tecnica consiste nella possibilità di inserire sulla superficie delle nanoparticelle gruppi chimici con affinità specifica verso recettori presenti in particolari tipi di cellule. Si possono così indirizzare le nanoparticelle, che grazie alle dimensioni ridotte sono in grado di circolare nei liquidi corporei, verso determinate cellule o tessuti-bersaglio, come vedremo meglio più avanti a proposito degli addotti polimerici di farmaci. Inoltre, è attualmente allo studio l’impiego di nanoparticelle opportunamente preparate per fungere da vettori non virali di acidi nucleici in esperimenti di trasfezione, o per altre applicazioni biotecnologiche.
Polimeri solubili come veicolantidi farmaci
Attualmente lo studio dei derivati polimerici di farmaci è certamente uno dei più affascinanti esempi di ricerca interdisciplinare, in cui convergono discipline diverse ma fra loro complementari, quali chimica, biologia, fisiologia e farmacologia. Si tratta di un campo relativamente recente, che quindi non ha ancora espresso pienamente tutto il suo potenziale in termini di applicazioni pratiche.
Le opportunità offerte dall’uso dei veicolanti polimerici solubili di farmaci derivano da due caratteristiche tipiche della loro natura: le dimensioni molecolari, superiori di due o tre ordini di grandezza rispetto a quelle delle molecole tradizionali, e la multifunzionalità, cioè il fatto che i polimeri, essendo per definizione costituiti da un numero molto grande di unità connesse fra loro, possono svolgere numerose funzioni chimiche per ogni molecola. La multifunzionalità comporta la possibilità di agganciare a ogni singola molecola di veicolante polimerico non soltanto numerose molecole di farmaco, ma anche altri gruppi chimici capaci di coadiuvarne l’attività. La catena polimerica, infatti, può essere assimilata a un filo cui è possibile appendere oggetti di diversa natura, ciascuno dei quali contribuisce con la propria funzione al conseguimento di uno scopo complessivo.
Differenze tra farmaci tradizionali e addotti polimerici di farmaci
Nei sistemi biologici le sostanze polimeriche e non polimeriche si comportano in modo differente. I polimeri somministrati per iniezione sono bioeliminabili solo quando il loro peso molecolare è inferiore a un valore soglia, che è solitamente di qualche decina di migliaia; polimeri di peso molecolare superiore sono bioeliminabili solo se il loro peso molecolare si riduce nel tempo in seguito a degradazione, mentre un polimero a elevato peso molecolare oltrepassa con difficoltà le pareti del tratto gastrointestinale e di regola non è significativamente assorbito dopo assunzione orale.
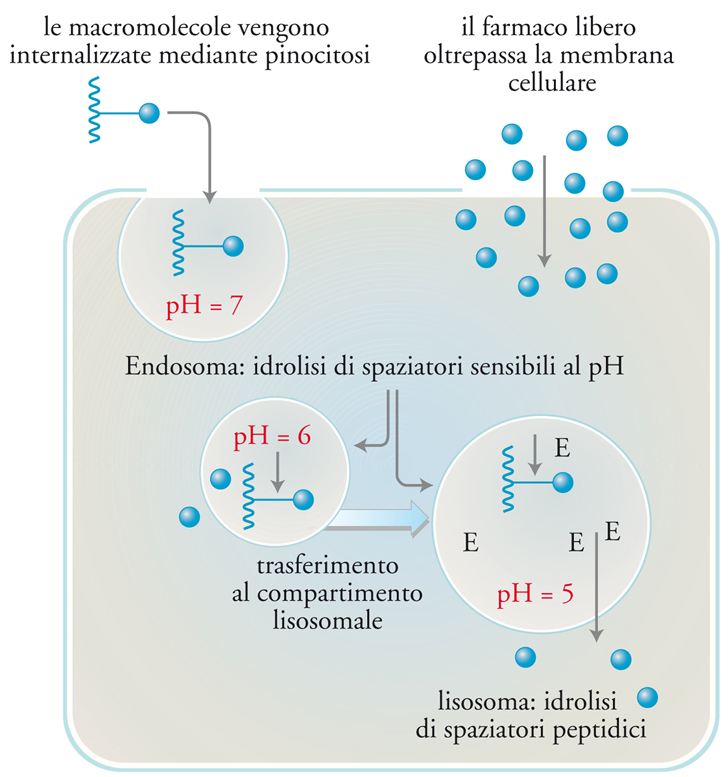
Inoltre, esistono differenze specifiche tra farmaci tradizionali e addotti polimerici di farmaci, e proprio su tali differenze si basa l’introduzione di questi ultimi come agenti antitumorali. I farmaci tradizionali trovano rapidamente accesso alle cellule passando attraverso le membrane cellulari. Nel caso della somministrazione per via endovenosa, un’elevata percentuale del farmaco iniettato viene di regola escretata dopo pochi minuti, mentre il resto si distribuisce omogeneamente nell’organismo. Invece, l’ancoraggio di un farmaco a un polimero ne altera profondamente la farmacocinetica, perché le elevate dimensioni molecolari impediscono la diffusione attraverso le membrane cellulari. L’internalizzazione dei polimeri nelle cellule avviene con un meccanismo diverso − chiamato con termine generale endocitosi, più specificamente nel caso considerato pinocitosi − consistente essenzialmente in un’invaginazione della membrana cellulare con creazione di un vacuolo (endosoma) che contiene una porzione del liquido intracellulare con tutte le sostanze in esso disciolte (fig. 16). Introducendo nel veicolante gruppi chimici con affinità specifica per i recettori complementari presenti sulla membrana di un certo tipo di cellula, l’addotto polimero-farmaco si concentrerà sulla membrana e quel tipo di cellula lo assumerà specificamente.
L’internalizzazione cellulare mediante endocitosi determina il rilascio dell’addotto negli endosomi, dove il pH scende fino a 6 e di qui successivamente nei compartimenti lisosomali, dove il valore del pH è 5,5. All’interno dei lisosomi il farmaco polimerico incontra le idrolasi lisosomali, enzimi in grado di scindere numerosi legami chimici. Questi cambiamenti nelle caratteristiche fisico-chimiche dell’ambiente in cui il farmaco si viene a trovare permettono di progettare legami farmaco-polimero in grado di essere degradati intracellularmente e con velocità di degradazione controllabile.
Direzionamento passivo e attivo verso i tumori
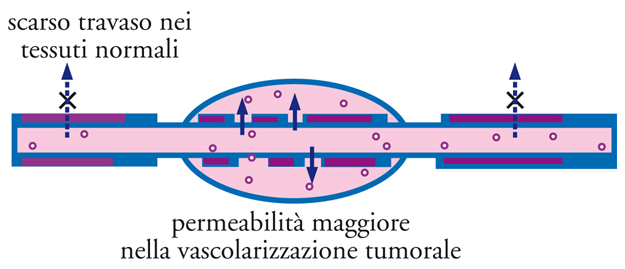
Si è scoperto che composti polimerici naturali o sintetici, quali l’albumina, gli addotti polimerici di farmaci e gli addotti fra polimeri e proteine, sono accumulati passivamente nei tessuti tumorali. Questo fenomeno è stato chiamato effetto EPR (Enhanced permeability and retention, aumentata permeabilità e ritenzione) ed è attribuibile a due principali fattori: da un lato, la vascolarizzazione tumorale mostra un endotelio discontinuo, cioè con permeabilità alterata, che ne permette l’attraversamento da parte di macromolecole di grosse dimensioni, le quali invece attraversano con difficoltà o non attraversano affatto le pareti di vasi normali (fig. 17); dall’altro lato, si osserva nel tumore la mancanza di un efficace drenaggio da parte del sistema linfatico, che conduce a un accumulo locale delle macromolecole penetrate nel tessuto.
Perché l’effetto EPR si esplichi pienamente, è necessario che la sostanza polimerica permanga a lungo nel circolo sanguigno prima di essere intercettata dai sistemi di difesa dell’organismo. Polimeri con queste caratteristiche sono definiti stealth, un termine col quale nel linguaggio militare vengono indicati gli aerei non intercettabili dai radar.
Probabilmente, dal punto di vista quantitativo, l’accumulo di addotti polimerici stealth nei tessuti tumorali è dovuto principalmente all’effetto EPR, ma non esclude la possibilità di sovrapporvi un direzionamento attivo mediato da recettori. Esistono efficaci agenti direzionanti, come il galattosio, specifico per gli epatociti, che è stato ampiamente utilizzato, per esempio, per addotti polimerici del farmaco antitumorale doxorubicina.
Elementi di progettazione di addotti polimerici di farmaci
Un veicolante polimerico ideale per la preparazione di addotti polimerici di farmaci deve avere una serie di requisiti, alcuni dei quali specifici, altri comuni a tutti i biomateriali: deve essere biocompatibile e bioeliminabile; deve possedere gruppi funzionali che permettano l’aggancio di una quantità sufficiente sia di farmaco, sia di altri gruppi chimici coadiuvanti; deve possedere un adeguato potere solubilizzante nel caso di farmaci poco solubili in acqua; deve accumularsi selettivamente a livello del sito d’azione preferenziale; deve provvedere un aggancio polimero-farmaco stabile nel sistema vascolare, ma degradabile nel sito d’azione preferenziale; deve, infine, essere riproducibile su scala industriale e facilmente caratterizzabile.
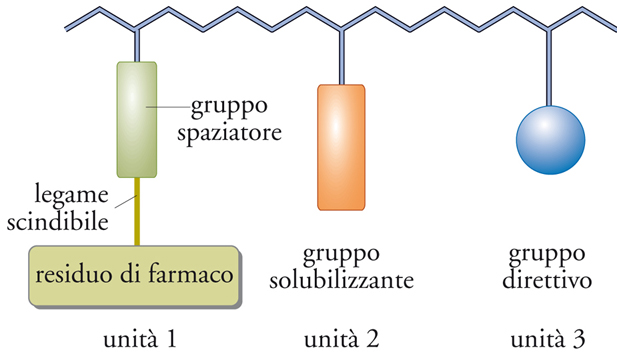
Gli elementi fondamentali degli addotti polimerici di farmaci sono codificabili in un modello generale, detto modello di Ringsdorf, illustrato in fig. 18. Nella figura viene mostrato un polimero in cui sono presenti tre diversi tipi di unità: un’unità cui è legato il farmaco tramite uno spaziatore; un’unità solubilizzante; un’unità contenente un gruppo avente la funzione di guidare il farmaco verso un recettore specifico. Il modello vale per ogni tipo di farmaco, ma è stato sviluppato soprattutto per i chemioterapici antitumorali, dei quali in teoria assicura la solubilizzazione, il direzionamento e la liberazione a livello del tumore. Questo modello è stato applicato nella preparazione di numerosi antitumorali polimerici che attualmente si trovano a vari stadi di sperimentazione clinica.
Bibliografia
Black 1999: Black, Jonathan, Biological performance of materials: fundamentals of biocompatibility, 3. ed., New York, Dekker, 1999.
Chiellini 2001: Biomedical polymers and polymer therapeutics, edited by Emo Chiellini e altri, New York-London, Klu-wer Academic, 2001.
Lanza 2000: Principles of tissue engineering, edited by Robert P. Lanza, Robert Langer, Joseph P. Vacanti, 2. ed., London-San Diego, Academic Press, 2000.
Ottenbrite, Kim 2001: Polymeric drugs & drug deliverysystems, edited by Raphael M. Ottenbrite, Sang Wan Kim, Lancaster (Penn.), Technomic, 2001.
Ratner 1996: Biomaterials science: an introduction to materials in medicine, edited by Buddy D. Ratner e altri, London-San Diego, Academic Press, 1996.
Ratner, Bryant 2004: Ratner, Buddy D. - Bryant, Stephanie, Biomaterials: Where we have been and where are we going, “Annual reviews of biomedical engineering”, 6, 2004, pp. 1-75.
Saltzman 2001: Saltzman, W. Mark, Drug delivery: engineering principles for drug therapy, Oxford, Oxford University Press, 2001.
Whittaker, Fillinger 2006: Whittaker, David R. - Fillinger, Mark F., The engineering of endovascular stent technology.A review, “Vascular and endovascular surgery”, 40, 2006, pp. 85-94.