astrocitoma
astrocitoma
Neoplasia che deriva dalle cellule astrocitarie, costituenti la macroglia (➔ glia). Le neoplasie astrocitarie sono di vario tipo e rientrano in due categorie: le forme localizzate, con prognosi relativamente favorevole, che comprendono l’a. pilocitico e lo xantoastrocitoma pleomorfo, e le forme diffusamente infiltranti che tendono a infiltrare il tessuto cerebrale anche a distanza e a progredire nel tempo verso gradi maggiori di malignità; queste comprendono l’a. diffuso, l’a. anaplastico ed il glioblastoma.
Astrocitoma pilocitico
Questo a. insorge tipicamente nei bambini e nei giovani adulti ed è localizzato in genere nel cervelletto ma può insorgere in altre zone dell’SNC quali il III ventricolo, le vie ottico-chiasmatiche e, meno frequentemente, nei gangli della base o nel midollo spinale. Nei pazienti affetti da neurofibromatosi (➔) di tipo 1 (nf1) è la forma più frequente di neoplasia cerebrale e si localizza prevalentemente nelle vie ottico-chiasmatiche. Macroscopicamente si presenta come una lesione cistica con nodulo murale o come una massa ben circoscritta. Istologicamente risulta composto da cellule allungate prive di evidenti atipie. Tra le cellule neoplastiche si osservano frequentemente fibre di Rosenthal e corpi granulosi. Gli a. pilocitici sono neoplasie a lenta crescita; la loro prognosi è eccellente, soprattutto dove è possibile una resezione chirurgica radicale, come nel cervelletto, mentre in localizzazioni chirurgicamente meno aggredibili, con ridotte possibilità di asportazione radicale, possono dare luogo a recidive. Alcuni studi genetici hanno dimostrato la presenza, nella grande maggioranza di a. pilociticidi, di un gene di fusione tra i geni kiaa1549 e braf che si esprime a livello cromosomico con una replicazione del cromosoma 7q34.
Xantoastrocitoma pleomorfo
Anche lo xantoastrocitoma pleomorfo è un tumore che colpisce bambini e giovani adulti. Insorge superficialmente, spesso interessando il lobo temporale con una storia di epilessia di lunga durata. All’esame microscopico è costituto da cellule astrocitarie marcatamente pleomorfe, cioè con grandi varietà di forme. con aspetti di lipidizzazione citoplasmatica. L’elevata cellularità e le atipie citologiche possono erroneamente indurre una diagnosi di malignità. La sua prognosi è in genere buona, con sopravvivenze dell’80% a cinque anni. La necrosi e le mitosi, quando presenti, indicano forme che possono avere un decorso più aggressivo.
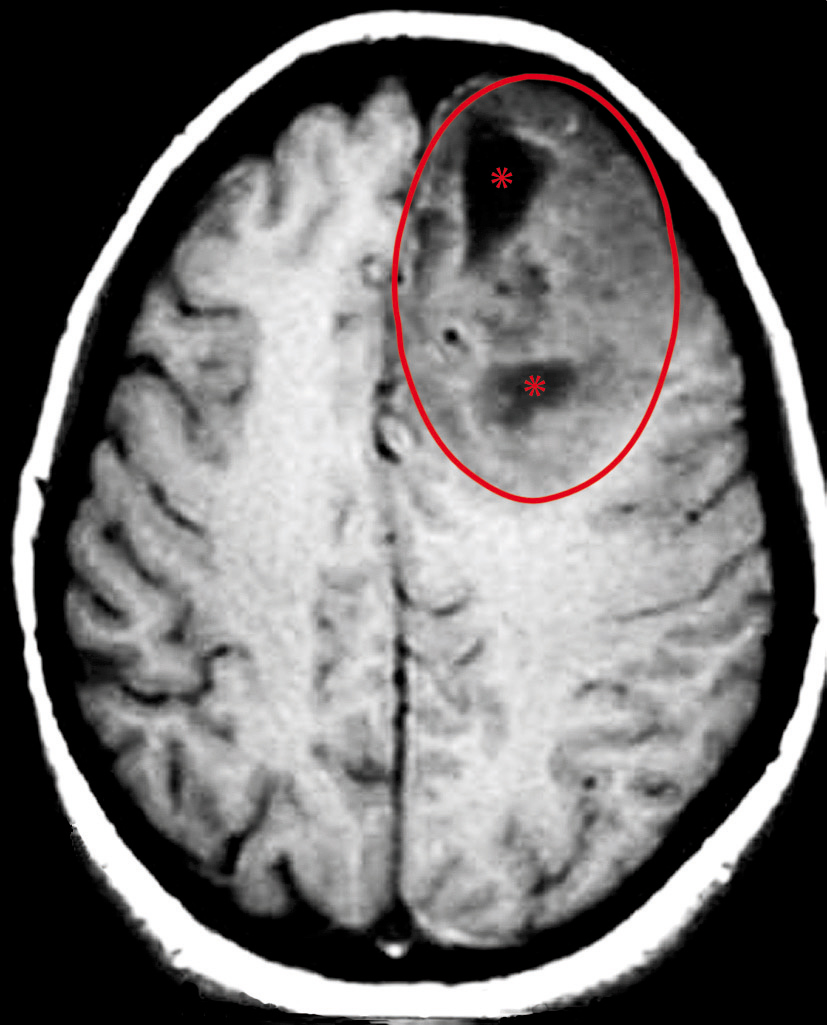
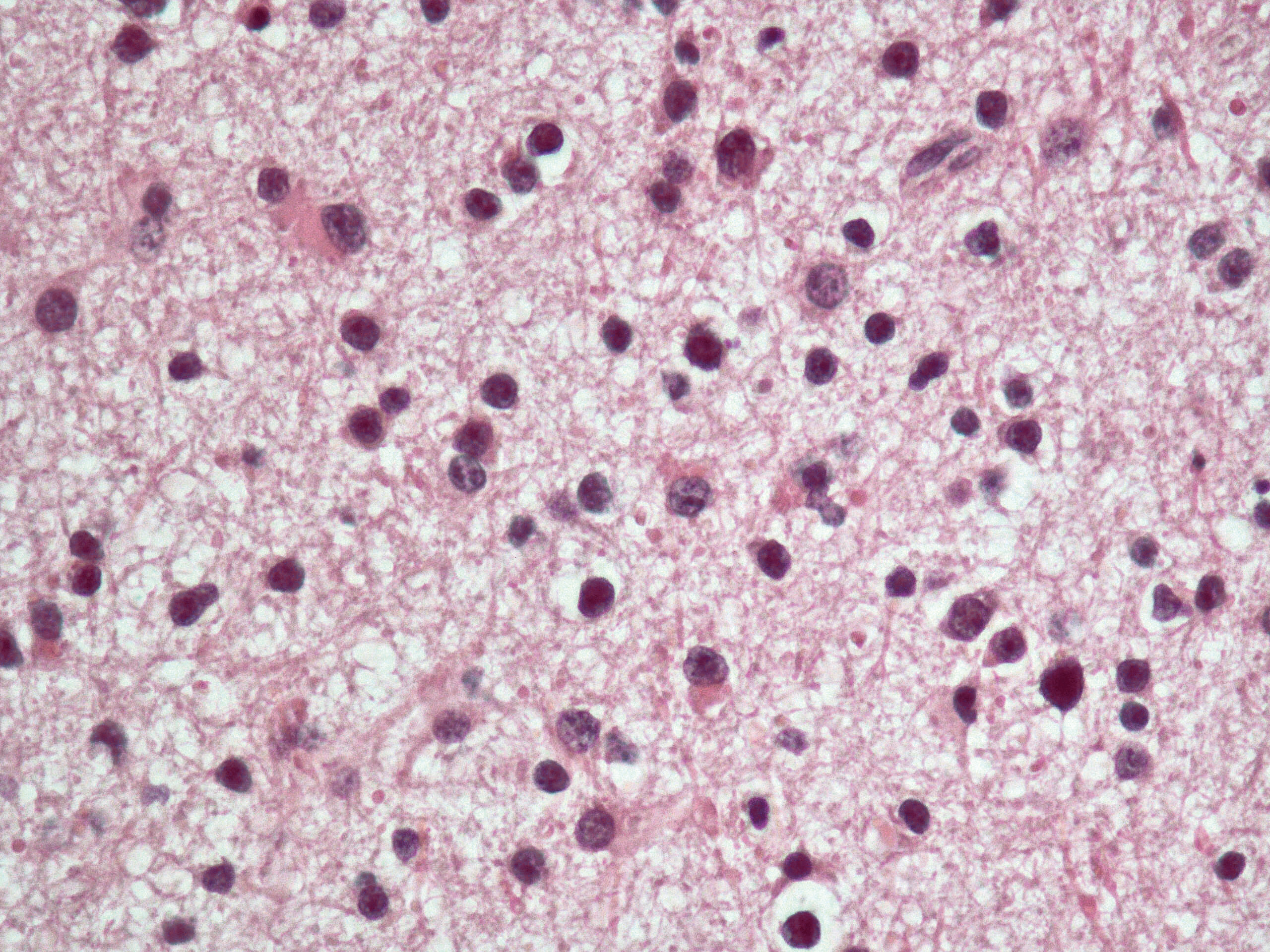
Astrocitomi diffusi e glioblastoma
Questo gruppo, che da solo rappresenta quasi l’80% delle neoplasie primitive dell’adulto, mostra uno spettro progressivo di malignità che varia, da forme ben differenziate di a. diffuso, di grado II (in base alla classificazione dell’Organizzazione mondiale della sanità, OMS), a forme meno differenziate ad alto grado quali l’a. anaplastico (grado III OMS) e il glioblastoma (grado IV OMS). L’a. diffuso (grado II) insorge in adulti con età compresa tra i 20 ed i 30 anni. Colpisce prevalentemente gli emisferi cerebrali ma può insorgere nel tronco cerebrale o nel midollo spinale. Macroscopicamente ingrandisce e distorce le strutture colpite, rendendone spesso sfumati i limiti anatomici. Istologicamente ha una cellularità bassa e le cellule neoplastiche mostrano moderato pleomorfismo nucleare. Le mitosi sono molto rare e non vi è proliferazione vascolare né necrosi. Le cellule neoplastiche infiltrano anche a distanza il tessuto nervoso circostante. L’ a. gemistocitico costituisce una variante in cui le cellule hanno un abbondante citoplasma eosinofilo e nuclei dislocati alla periferia. Le alterazioni molecolari caratteristiche dell’a. diffuso di grado II sono la mutazione della proteina p53 dimostrata in circa il 50% dei casi, iperespressione di PDGf (Platet-Derived Grouth Factor) e del suo recettore PDGfR e perdita di porzioni del cromosoma 22. La maggioranza di questi a. di basso grado progrediscono verso forme ad alto grado nell’arco di 4÷5 anni in media. L’a. anaplastico (grado III OMS) spesso si sviluppa da a. diffusi di basso grado; l’età media di insorgenza è di circa una decade in più di quella del grado II. Istologicamente presenta una maggiore cellularità, un più marcato pleomorfismo nucleare e mitosi. Tuttavia necrosi e proliferazione vascolare sono assenti. A livello molecolare si osserva inattivazione dei geni che controllano il ciclo cellulare quali cdkn2a/p16/arf, cdk4, e del suo recettore rb, e presenta anche perdita di parti del braccio lungo del cromosoma 19. Gli a. anaplastici sono neoplasie aggressive con sopravvivenze da 2 a 3 anni dalla diagnosi. Il glioblastoma (➔) è la forma più frequente di neoplasia primitiva cerebrale ed è anche la forma più maligna di neoplasia astrocitaria.