
Apoptosi: morte cellulare programmata
Apoptosi: morte cellulare programmata
L'essere umano è un organismo pluricellulare in cui la divisione cellulare avviene mediante mitosi. Durante la mitosi, le cellule duplicano il proprio DNA e si dividono per formare due cellule figlie, ciascuna delle quali è la replica esatta della cellula genitrice. Nonostante in un organismo pluricellulare come quello umano gli eventi mitotici si verifichino costantemente in numero considerevole, le cellule del nostro corpo non vanno mai incontro ad alcuna variazione effettiva di numero. Ciò naturalmente implica che per ogni cellula generata con la mitosi, una deve morire; una cellula può morire attraverso due processi diversi, ossia la necrosi e l'apoptosi.
La necrosi è la forma di morte cellulare nella sua definizione classica ed è la conseguenza di un grave trauma subito dalla cellula. Una cellula che muore per necrosi va incontro a un rigonfiamento rapido e incontrollato e alla fine scoppia. Il suo contenuto intracellulare fuoriesce nell'ambiente extracellulare dando luogo, molto spesso, a una risposta infiammatoria, caratterizzata dall'aumento del flusso sanguigno nell'area interessata, dall'afflusso dei leucociti e dal rilascio di varie molecole che mediano la risposta infiammatoria. La morte cellulare per necrosi si osserva raramente in condizioni fisiologiche e le cellule che muoiono a causa di questo processo non hanno alcun controllo sul proprio destino.
Il secondo tipo di morte cellulare, detto 'apoptosi', è stato scoperto piuttosto recentemente. Al contrario della necrosi, l'apoptosi è un processo di morte cellulare controllato geneticamente. Nel 1972 Andrew H. Wyllie pubblicò insieme a John F. Kerr e Alastair R. Currie un importante articolo in cui si descriveva il processo di morte cellulare per apoptosi. Questo termine, di derivazione greca, è utilizzato per descrivere la caduta dei petali dei fiori o quella delle foglie dagli alberi. Infatti, le cellule che muoiono per apoptosi si condensano e si staccano letteralmente dalle strutture di supporto tissutali sulle quali stanno crescendo. Il processo di apoptosi fu identificato nel 1972; tuttavia, prima di allora la letteratura ne aveva riportato molti esempi: le cellule apoptotiche del timo (tingle bodies), i cheratinociti apoptotici della pelle (sunburn cells) e le cellule apoptotiche del fegato (councilman bodies) sono tutti esempi di apoptosi descritti in anni antecedenti il 1972. Un altro termine utilizzato per descrivere il fenomeno di apoptosi prima dello stesso anno è stato shrinkage necrosis, necrosi per riduzione di volume. Questa definizione costituisce un'accurata descrizione istologica dell'apoptosi, dato che la fuoriuscita di acqua è una delle sue caratteristiche chiave.
Nella seconda metà degli anni Ottanta gli immunologi e gli embriologi riscoprirono il lavoro di Wyllie e dei suoi collaboratori e iniziarono a esplorare i meccanismi biologici alla base dell'apoptosi sia nel sistema immunitario sia nell'ontogenesi. La ricerca tesa a scoprire perché e in che modo una cellula mette in moto il suo programma di morte cellulare ha costituito una delle aree più attive della biologia degli anni Novanta: la comprensione del processo mediante il quale una cellula si uccide potrebbe permettere di prolungarne la vita o di accelerarne la morte, un traguardo ambito in diverse patologie umane.
Sommario: 1. La morte cellulare durante lo sviluppo. 2. Caenorhabditis elegans e apoptosi. 3. Stress cellulare e apoptosi. 4. Caratteristiche biologiche dell'apoptosi. 5. Geni e apoptosi. 6. Enzimi proteolitici e apoptosi. 7. Meccanismo molecolare dell'apoptosi: l'apoptosoma. 8. Morte cellulare nel sistema immunitario. 9. Morte cellulare nel sistema nervoso. □ Bibliografia.
La morte cellulare durante lo sviluppo
Prima di tutto è utile chiedersi perché le cellule abbiano sviluppato un meccanismo sofisticato per morire e in quali circostanze venga attivato il programma di morte cellulare. Durante lo sviluppo fetale, per esempio, tra le dita è presente una rete di cellule che conferisce a queste strutture anatomiche l'aspetto di una pinna. Con il progredire dello sviluppo da questa pinna si modellano le dita e le cellule interdigitali muoiono per apoptosi. Questo è un chiaro esempio di come il corpo utilizzi il processo apoptotico di morte geneticamente programmata per formare nuove strutture anatomiche. Un altro esempio del ruolo svolto dall'apoptosi si ritrova nell'ontogenesi del sistema nervoso. Di fatto circa il 90% di tutti i neuroni formatisi durante la vita muore per apoptosi; ciò avviene perché essi non riescono a creare con i neuroni vicini connessioni che possano mantenerli in vita. Tale tipo di processo in cui si attua la connessione oppure si verifica la morte neuronale è di vitale importanza in quanto assicura che il sistema nervoso, il quale dipende strettamente dalla formazione corretta delle connessioni tra le cellule, sia assemblato con estrema cura per formare una rete neuronale funzionante. Un ultimo esempio è il riassorbimento della coda di un girino durante la sua trasformazione in rana.
Altri esempi provengono dall'immunologia nella quale le cellule autoreattive del sistema immunitario vengono eliminate prima che possano nuocere all'organismo, determinando una risposta immunitaria indesiderata a danno dei tessuti normali dell'individuo. Tuttavia, esistono diverse situazioni in cui la regolazione dell'eliminazione delle cellule del sistema immunitario è compromessa provocando l'insorgenza di una serie di malattie autoimmuni molto debilitanti.
Caenorhabditis elegans e apoptosi
L'organismo utilizzato preferenzialmente come modello per studiare una varietà di processi ontogenetici, in particolare l'apoptosi, è il verme nematode Caenorhabditis elegans. Questo organismo è composto di 1090 cellule di cui 131 muoiono in modo programmato per apoptosi, e ciò lo rende particolarmente interessante. La morte cellulare colpisce principalmente il sistema nervoso (105 cellule). Il ruolo centrale nella morte di queste 131 cellule è svolto da due geni chiamati ced-3 e ced-4, i quali vengono attivati selettivamente quando viene avviato il programma di apoptosi. Se questi geni mutano, tutte le 131 cellule destinate a morire sopravvivono, ma i vermi appaiono normali. Il riassorbimento e la rimozione delle cellule morte per apoptosi vengono controllati da altri sette geni che però non hanno alcun legame diretto con i due precedenti.
Lo scopo principale della morte cellulare in C. elegans è l'eliminazione delle cellule indesiderate e ciò, per esempio, può essere utile per eliminare le cellule che hanno eseguito un compito specifico durante i primi stadi dello sviluppo, ma che non hanno altre funzioni da svolgere nell'organismo adulto. In secondo luogo, può servire a generare il dimorfismo sessuale. Molti dei geni identificati in questo organismo e implicati nella regolazione dell'apoptosi hanno un corrispettivo negli organismi superiori quali i Mammiferi. Nel 2002 il Premio Nobel per la medicina o la fisiologia è stato attribuito, in compartecipazione, a Robert Horvitz per il raffinato lavoro volto a chiarire alcuni aspetti della genetica dell'apoptosi in C. elegans.
Stress cellulare e apoptosi
Gli organismi, nel corso della loro vita, sono soggetti a una varietà di stress naturali. Negli organismi pluricellulari questa condizione sussiste anche a livello cellulare. Se una cellula è soggetta a moderati livelli di stress, la sintesi di un gruppo di proteine da stress o da shock termico, o heat-shock proteins, può fornire un certo grado di protezione alla cellula, sempre che l'agente stressante venga eliminato dopo un breve periodo di tempo. Le proteine da stress si rinvengono in quasi tutti i tipi di cellule, dai batteri agli esseri umani, ed è probabile che costituiscano un sistema primitivo di autodifesa. Tuttavia, se la cellula è sottoposta a un insulto, insufficiente a ucciderla subito, ma dal quale le heat-shock proteins non possono proteggerla, allora viene attivato il programma di apoptosi ed essa muore in modo controllato (fig. 2). Ciò costituisce un grande vantaggio per gli organismi pluricellulari, perché, quando una cellula muore per apoptosi, il suo contenuto non è riversato all'esterno, come avviene invece nel processo di necrosi. In altre parole, con la morte per apoptosi una cellula compie l'atto 'altruistico' di salvare le cellule adiacenti. Infine, se la cellula subisce un insulto così grave da non avere il tempo di attivare il programma di apoptosi, la morte avviene allora per necrosi con danni e lesioni ben distinti alle cellule adiacenti che possono anche dar luogo alla morte delle altre cellule o dell'organismo stesso.
Caratteristiche biologiche dell'apoptosi
Una delle peculiarità biochimiche più tipiche dell'apoptosi è la rottura del DNA cellulare in frammenti delle dimensioni di un nucleosoma che, dopo elettroforesi su gel di agarosio, appaiono come i pioli di una 'scala' (DNA ladder). La frammentazione si compie con un processo in due stadi: inizialmente il DNA è suddiviso in grandi pezzi, i quali vengono ridotti in frammenti delle dimensioni di un nucleosoma nel passaggio successivo. È probabile che nel processo descritto intervengano due distinti sistemi enzimatici, in quanto alcune cellule sono capaci di produrre soltanto i frammenti grandi, mentre altre possono produrre entrambi i tipi.
Durante l'apoptosi la morfologia della cellula morente va incontro a una serie di cambiamenti consistenti, uno dei quali è la riduzione di volume dovuta all'espulsione di acqua (il volume cellulare può ridursi fino al 50%), ma non sappiamo quasi niente dei motivi per cui la cellula vada soggetta a questo restringimento o dei modi in cui esso si attui. Oltre a questo fenomeno, nella cellula si verifica anche una marcata condensazione della cromatina che origina i nuclei picnotici. In alcune cellule, il nucleo va poi incontro a frammentazione all'interno della cellula. Il successivo cambiamento morfologico evidente è la rottura della cellula in piccole vescicole chiuse chiamate corpi apoptotici. Le vescicole vengono poi inglobate e distrutte dai fagociti circostanti. Durante l'intero processo, il contenuto cellulare non fuoriesce nell'ambiente extracellulare, in quanto si conserva l'integrità della membrana plasmatica.
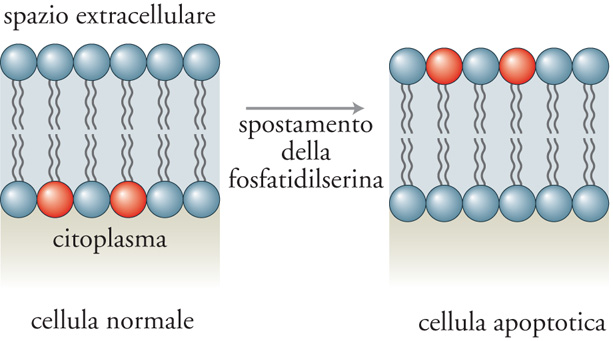
Uno dei motivi per cui gli scienziati hanno impiegato tanto tempo a scoprire l'apoptosi è da imputarsi alla rapidità con la quale in condizioni fisiologiche le cellule apoptotiche vengono rimosse. Il riconoscimento e la rimozione delle cellule apoptotiche sembrano correlati all'espressione di nuove molecole sulla loro superficie, che ne consentono l'identificazione da parte dei fagociti. Una di queste molecole è il lipide di membrana fosfatidilserina, normalmente localizzato sulla superficie interna della membrana plasmatica, il quale durante l'apoptosi migra verso la superficie esterna agendo come segnale di riconoscimento per la fagocitosi (fig. 3).
Geni e apoptosi
Grazie al lavoro pubblicato negli anni Ottanta da J. John Cohen e Rick C. Duke (1984) sappiamo che l'apoptosi è un processo controllato geneticamente. Esperimenti successivi hanno condotto all'identificazione di due classi di geni coinvolti nella regolazione dell'apoptosi. In una classe si trovano geni quali c-myc e p53 che dirigono il processo apoptotico, nell'altra vi sono geni come bcl-2 e bcr-abl che possono inibire la morte cellulare.
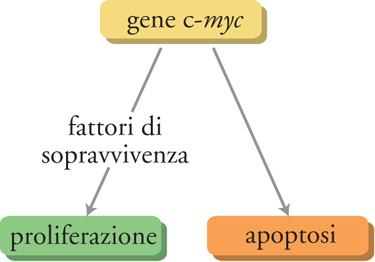
Il gene c-myc è considerato una sorta di 'Giano bifronte' in quanto può guidare sia la proliferazione sia la morte delle cellule. Per esempio, in presenza dei fattori di sopravvivenza cellulare come il fattore di crescita insulinosimile-1 (IGF-1), l'espressione del gene c-myc induce la proliferazione; invece, in assenza di tali fattori il gene c-myc causa la morte cellulare per apoptosi. C-myc è un gene chiave nella regolazione della proliferazione cellulare e perciò ogni mutazione che ne amplifichi l'espressione ha la potenzialità di portare a una proliferazione cellulare incontrollata e al possibile sviluppo di tumori. Tuttavia c-myc non controlla da solo la proliferazione cellulare: deve essere stata prodotta un'adeguata quantità di fattori di sopravvivenza cellulare, la cui sintesi è regolata da altri geni che vengono attivati soltanto quando è necessario. Perciò anche in presenza di un aumento incontrollato dell'espressione di c-myc, il tumore non si sviluppa a meno che non si verifichi un altrettanto incontrollato aumento dell'espressione dei fattori di sopravvivenza. In altre parole, perché si sviluppi una cellula tumorale devono essere mutati almeno due geni chiave e ciò probabilmente spiega, in parte, perché la conversione di una cellula normale in una tumorale sia un evento relativamente raro, considerata l'elevata frequenza delle divisioni cellulari normali che si verificano nel nostro corpo (fig. 4).
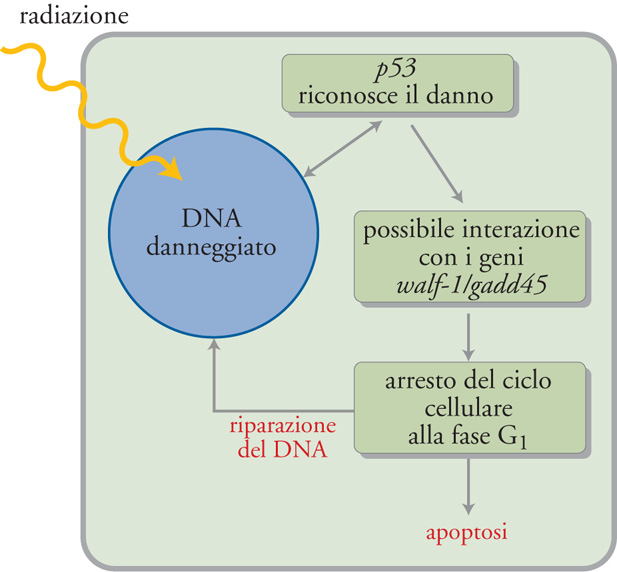
Un altro gene chiave che favorisce l'apoptosi e che in determinate circostanze causa la morte cellulare è p53, che risulta mutato nella grande maggioranza dei tumori umani. Per questo motivo esso fa parte di una famiglia di geni, noti come 'soppressori tumorali', che, quando sono attivi, impediscono lo sviluppo dei tumori. La funzione principale di p53 oggi nota è di bloccare le cellule che hanno subito un danno a livello del DNA nella fase G1. Tale blocco permette alla cellula di riparare il DNA e, se la riparazione non è possibile, viene avviato il processo di apoptosi. Se p53 non riesce a svolgere i suoi compiti, per esempio a causa di mutazioni, allora le cellule con il DNA danneggiato possono rientrare nel ciclo cellulare con la possibilità di dare origine al cancro. Il gene p53 quando è mutato non può dare avvio all'apoptosi. Non è chiaro come p53 medi i propri effetti, ma la sua capacità di bloccare le cellule in G1 viene probabilmente realizzata attraverso l'interazione con altri geni, quali waf-1 e gadd45, noti per il loro coinvolgimento nel controllo del ciclo cellulare (fig. 5). Per il suo ruolo di vigilanza sul ciclo cellulare, il gene p53 è stato soprannominato 'il custode del genoma'.
Proprio come esistono geni che favoriscono l'apoptosi, quali c-myc e p53, parimenti esiste una classe di geni che inibisce l'apoptosi; in questa categoria rientrano geni come bcl-2 e bcr-abl. Il primo oncogene di cui si è identificato un ruolo di controllo nel processo di apoptosi è stato bcl-2; le cellule in cui questo gene viene espresso a livelli elevati sono considerevolmente più resistenti all'induzione di apoptosi. È stato dimostrato che l'espressione del gene bcl-2 è associata al linfoma delle cellule follicolari e ciò ha suggerito che questo tipo di cancro possa essere dovuto più a una mancanza di morte cellulare che a un eccesso di proliferazione. Un'altra conseguenza dell'elevata espressione di bcl-2 è una maggiore resistenza delle cellule ai farmaci. Lo scenario a nostra disposizione mostra perciò l'esistenza di un gene che, inibendo la morte cellulare, non solo contribuisce allo sviluppo del tumore, ma rende anche più difficile uccidere le cellule tumorali con i convenzionali farmaci contro il cancro. L'espressione di bcl-2 sembra contribuire anche allo sviluppo e alla progressione di altri tipi di cancro, come quello della prostata.
I pazienti affetti da questa patologia hanno in genere difficoltà a urinare a causa dell'ipertrofia della ghiandola prostatica. Il cancro della prostata può caratterizzarsi come una malattia bifasica, in cui la fase iniziale risponde al trattamento. Durante questa fase le cellule prostatiche cancerose proliferano in risposta agli androgeni, come il testosterone, che favoriscono la crescita cellulare; se gli androgeni vengono eliminati, il tumore si rimpicciolisce notevolmente e le condizioni del paziente migliorano sensibilmente. Ciò può essere ottenuto usando sostanze che antagonizzano gli effetti degli androgeni o con la chirurgia, che nella forma della castrazione può essere utilizzata per rimuovere la fonte degli ormoni. Purtroppo, come altri tipi di cancro, anche quello della prostata inevitabilmente recidiva, con una seconda fase assai più resistente al trattamento e che di solito è fatale. In questa fase, infatti, il cancro recidivante è indipendente dagli androgeni e quando è eliminato l'ormone che ne favorisce la crescita le cellule non muoiono per apoptosi. Nelle cellule tumorali che rispondono all'ormone, l'espressione di bcl-2 è minima o assente, mentre è presente quella del gene bax, che favorisce la morte cellulare. Quando lo stimolo proliferativo degli androgeni viene rimosso, chimicamente o con la castrazione, le cellule muoiono in breve tempo. Viceversa, bcl-2 previene questa morte e le cellule tumorali sopravvivono. È probabile che l'espressione di questo gene sia coinvolta nel passaggio del cancro da una fase che inizialmente risponde bene alla terapia a una fase resistente. Tale trasformazione non si osserva soltanto nel cancro della prostata, ma anche in un numero crescente di altri tipi di cancro.
La famiglia delle proteine bcl-2 svolge un ruolo fondamentale nel controllare il destino delle cellule. Queste proteine sono regolatori chiave dell'apoptosi, anche se ancora non comprendiamo il meccanismo preciso attraverso cui esse svolgono le loro funzioni. In origine bcl-2 fu scoperto nei linfomi delle cellule B dove la sua espressione veniva amplificata. Ciò si traduceva nel blocco della normale apoptosi delle cellule B, nell'accumulo di queste cellule 'non morte' e nello sviluppo del tumore. Questa osservazione ha dimostrato che il tumore può insorgere anche in conseguenza del blocco della morte cellulare oltre che per un incremento della proliferazione. Da allora si è aggiunto a questa famiglia un numero sempre crescente di nuove proteine, di cui si può dire che bax sia la più conosciuta. Essa agisce di concerto con bcl-2 per promuovere la morte cellulare. Un'ipotesi di studio molto affascinante sostiene che la principale funzione di bcl-2 sia proprio quella di mantenere bax in uno stato inattivo (Oltvai et al. 1993). A seconda del bilancio tra queste due proteine una cellula può essere indirizzata verso la sopravvivenza o la morte.
Oltre a bcl-2 si sono dimostrati potenti inibitori dell'apoptosi anche bcr-abl e v-abl. Per esempio, la leucemia mieloide cronica è caratterizzata dalla traslocazione del gene c-abl dal cromosoma 7 al cromosoma 22. Ciò porta alla produzione della proteina di fusione bcr-abl. Il meccanismo mediante il quale bcr-abl e v-abl mediano i loro effetti non sembra coinvolgere l'azione di bcl-2 o bax, ma un altro sistema effettore ancora del tutto sconosciuto. La leucemia mieloide cronica è una malattia bifasica, la cui fase iniziale è caratterizzata dall'accumulo di un gran numero di cellule mature della serie mieloide. Ciò sembra dovuto a un difetto di apoptosi, probabilmente causato da un aumento dell'attività chinasica della proteina mutata bcr-abl. La malattia può rimanere in una fase cronica per settimane o anni ma è inevitabile che prima o poi avvenga la transizione a una forma più acuta. A livello biochimico si verifica una mutata espressione dei geni che regolano la proliferazione e questo, insieme alla presenza di geni che prevengono la morte cellulare, fornisce alle cellule tumorali tutti gli ingredienti per sopravvivere e proliferare. Una delle questioni biochimiche fondamentali è in che modo l'aumento dell'attività chinasica consenta ad Abl di sopprimere la morte cellulare. Una via di segnalazione chiave è l'attivazione della cascata della chinasi PI3/Akt che impartisce un forte segnale di sopravvivenza alla cellula. L'attivazione di questa via si rivela, dunque, assai importante per la sopravvivenza di molte cellule tumorali.
Enzimi proteolitici e apoptosi
È possibile dedurre il ruolo degli enzimi proteolitici da diverse osservazioni sperimentali, incluso il fatto che gli inibitori delle proteasi sembrano rallentare l'inizio dell'apoptosi. Una seconda osservazione che ha focalizzato le ricerche sul ruolo delle proteasi nell'apoptosi è stata che la sequenza di ced-3, è simile a quella dell'enzima convertitore della interleuchina 1β umana (ICE), che nell'uomo costituisce il mediatore dell'infiammazione. Questo enzima è stato inizialmente scoperto nei macrofagi, dove sembra tagliare la prointerleuchina I trasformandola nella sua forma attiva; inoltre appartiene a una famiglia di enzimi chiamati 'cisteina-proteasi'. Diversi laboratori iniziarono a cercare non soltanto altri enzimi simili a ICE, ma anche i loro substrati nell'apoptosi. Vennero scoperti molti enzimi di questo tipo ora denominati caspasi. A tutt'oggi sono stati identificati circa 14 membri di questa famiglia di enzimi caratterizzati dalla capacità di tagliare i loro substrati polipeptidici immediatamente dopo i residui di acido aspartico. Approssimativamente questi enzimi possono essere suddivisi in due gruppi, uno implicato nella fase iniziale dell'apoptosi e l'altro in quella di attuazione. La caspasi-3 è una classica caspasi implicata nella fase di attuazione che può essere attivata dalle caspasi-8 e -9, due enzimi classificabili come iniziatori. Una volta attivata, la caspasi-3 può spezzare una quantità di substrati proteici che portano al collasso e alla morte delle cellule. Tali substrati comprendono l'enzima di riparazione del DNA, la poliADP-ribosio-polimerasi (PARP). Un secondo bersaglio è costituito dalla proteina ICAD che inibisce l'endonucleasi del DNA chiamata CAD. Una volta rilasciato dal suo inibitore, questo enzima procede al taglio del DNA in frammenti delle dimensioni di un nucleosoma, i quali costituiscono la caratteristica più tipica dell'apoptosi.
Meccanismo molecolare dell'apoptosi: l'apoptosoma
Un fattore chiave dell'apoptosi in C. elegans è la molecola ced-4. Ebbene, nei Mammiferi, un solo gene sembra essere l'omologo di ced-4. Si tratta del gene codificante per il fattore di attivazione delle proteasi apoptotiche (Apaf1). Questo è piuttosto sorprendente dal momento che, in genere, aumentando la complessità dell'organismo, aumenta la complessità dell'organizzazione molecolare e, quindi, il numero di geni coinvolti. In effetti, per gran parte dei geni di C. elegans esiste nei Mammiferi, in qualità di controparte funzionale, un'intera famiglia genica con numerose componenti che svolgono funzioni parzialmente sovrapposte. Per esempio il gene ced-3 ha nei Mammiferi come controparte un'intera famiglia genica, codificante per le caspasi. Il fattore apoptotico Apaf1 ha grandi dimensioni (una massa molecolare di ben 135 kDa) e tre domini proteici di grande interesse. Il primo è il cosiddetto 'dominio CARD' (Caspase recruitment domain). Esso è comune a molte altre proteine, fra cui la caspasi-9 e, non a caso, l'interazione fra i domini CARD di Apaf1 e della caspasi-9 è molto importante per il funzionamento di entrambe le molecole. Il secondo è un dominio di legame a nucleotidi liberi (ATP/dATP, adenosintrifosfato/deossiadenosintrifosfato), mentre il terzo è un dominio del tipo WD-40, debolmente conservato in natura e generalmente molto rilevante nelle interazioni fra proteine.
In condizioni fisiologiche e in assenza di stimolo apoptotico, la molecola Apaf1 è presente nel citoplasma di gran parte delle cellule dell'organismo in una forma strutturalmente 'chiusa' e funzionalmente inattiva in cui la sua estremità amminica è legata alla sua estremità carbossilica. Quando uno stimolo apoptotico interno alla cellula (per es., la segnalazione da parte del nucleo di un problema grave e irreveresibile di danneggiamento del DNA) o esterno alla cellula (quale la segnalazione da parte di recettori posti sulla membrana plasmatica che comunicano un difetto di nutrienti o un preciso comando di induzione alla morte) raggiunge il mitocondrio, questo viene sconvolto da una serie di modificazioni significative. Fra queste la più importante è certamente il rilascio all'esterno di una proteina essenziale alla vita della cellula, il citocromo c. Questa molecola è generalmente impegnata nel trasporto di elettroni caratteristico della fosforilazione ossidativa, un processo che consente alla cellula di produrre diverse molecole di ATP, la principale fonte di energia per il metabolismo cellulare. Una volta rilasciato nel citoplasma, il citocromo c si lega rapidamente ad Apaf1 e ne induce una modificazione conformazionale che consente la sua 'apertura' strutturale e l'attivazione funzionale. Apaf1 in conformazione 'aperta' è infatti in grado di legare attraverso il suo dominio CARD una molecola di caspasi-9. In un perfetto rapporto stechiometrico 1:1 e in rapida successione, altre sei molecole di caspasi-9 vengono reclutate da altrettante molecole di Apaf1, fino alla formazione di un complesso multimolecolare eptamerico a forma di ruota, denominato apoptosoma, dotato di un fulcro centrale (costituito dai domini CARD di Apaf1 e caspasi-9) e sette raggi (costituiti dai domini WD-40 di Apaf1 legati ad altrettante molecole di citocromo c). Il nucleotide libero ATP/dATP, una volta legato ad Apaf1, contribuisce al consolidamento della sua conformazione aperta. Alla formazione dell'apoptosoma fa seguito l'attivazione della caspasi-9, che a sua volta promuove l'attivazione di caspasi esecutrici quali le caspasi -3, -6 e -7. In particolare, la caspasi-3 viene fisicamente reclutata sull'apoptosoma. Mediante un fenomeno di amplificazione retroattiva, la caspasi-3 è in grado di indurre un'attivazione alternativa delle molecole di caspasi-9 rimaste libere nel citoplasma, guidando la cellula al suo inesorabile destino verso la demolizione di tutte le strutture nucleari e citoplasmatiche.
Morte cellulare nel sistema immunitario
La sindrome da immunodeficienza acquisita (AIDS) è una malattia caratterizzata dal lento deterioramento del sistema immunitario, la cui causa sembra essere una massiccia diminuzione di un gruppo di linfociti T detti 'cellule CD4'. La funzione principale di queste cellule è di controllare e stimolare la risposta immunitaria alle infezioni. Sembra che la morte di tali cellule nei pazienti affetti da HIV, ossia dal virus dell'immunodeficienza umana, avvenga per apoptosi, sebbene gli eventi cellulari che provocano la morte cellulare siano ancora sconosciuti. Ciò che sappiamo con certezza è che, in gran parte, l'apoptosi delle cellule CD4 non è mediata direttamente dal virus dell'AIDS, ma attraverso un meccanismo indiretto. Una volta entrato nella cellula tramite gp120, una delle proteine strutturali del virus che riveste un ruolo fondamentale nel processo che gli consente di prendere contatto con i linfociti CD4, il virus si integra nel genoma cellulare dove rimane finché non è stimolato a replicarsi. La proteina gp120 viene prodotta anche durante il processo replicativo e può essere rilasciata nel siero dove può legarsi ad altre cellule. Se stimolate per dare origine a una risposta immunitaria, queste stesse cellule vanno incontro ad apoptosi invece di proliferare come farebbero in un processo di risposta normale. Perciò, quando una cellula CD4 riceve un segnale di proliferazione, se nello stesso tempo si è legata a una proteina gp120, si comporta esattamente al contrario in risposta ai segnali provenienti dalla proteina virale. Mentre la proteina gp120 e la stimolazione della cellula CD4 attraverso il suo recettore possono essere gli eventi chiave nella stimolazione dell'apoptosi, poco si sa riguardo al modo in cui avviene esattamente l'apoptosi delle cellule CD4. L'effettiva morte cellulare potrebbe essere causata da una molecola della superficie cellulare detta 'Fas' e dal suo ligando (FasL). Negli ultimi anni Fas è diventata una molecola particolarmente importante per la comprensione dei primi passaggi del meccanismo di apoptosi.
Fas appartiene alla famiglia dei TNF (Tumour necrosis factor) così chiamati per il loro ruolo nella morte cellulare. Essa è espressa sulla superficie di molte cellule del corpo e, quando si lega al suo ligando naturale, induce l'apoptosi. La maggior parte delle nostre conoscenze su Fas deriva dagli studi effettuati sulle cellule del sistema immunitario, dove questa proteina sembra svolgere un ruolo cruciale nell'apoptosi. Nel timo, organo responsabile dello sviluppo dei linfociti T, circa il 99% di tutti i timociti prodotti muore e sembra che la morte avvenga per apoptosi naturale. I timociti esprimono un elevato livello di Fas e vi sono numerose prove del fatto che la morte cellulare osservata in queste cellule sia mediata da Fas. Le mutazioni nella molecola Fas causano la proliferazione dei linfociti; ciò indica che questa molecola è fondamentale nella regolazione dell'equilibrio tra proliferazione e morte dei linfociti. Fas ha inoltre un ruolo nella distruzione delle cellule tumorali a opera dei linfociti T citotossici. In altri termini, i linfociti T citotossici uccidono le loro cellule bersaglio inducendo l'apoptosi; in questo processo sembra essere coinvolta anche la proteina Fas.
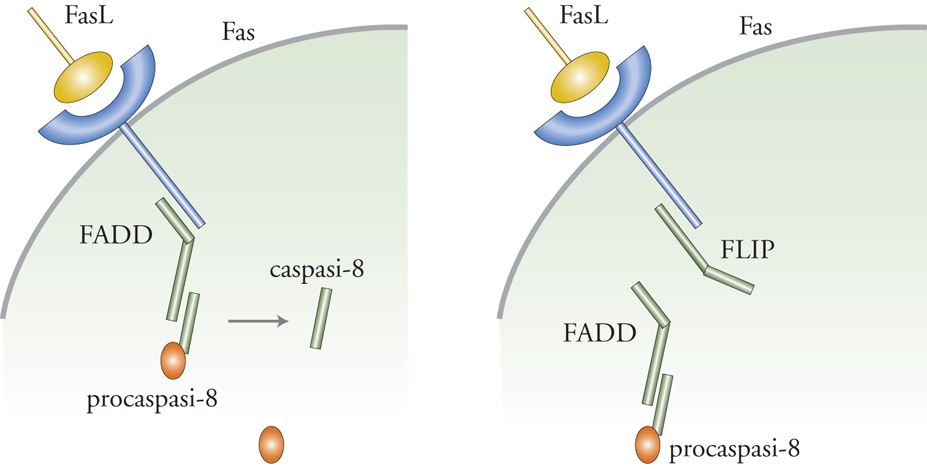
Fas è una proteina recettore transmembrana con una coda citoplasmatica contenente un cosiddetto 'dominio di morte'. Tale denominazione è dovuta al fatto che le mutazioni in questa regione della molecola ne bloccano la capacità di indurre la morte cellulare. Uno dei quesiti fondamentali che i ricercatori si pongono riguarda il modo in cui il recettore Fas determini le principali caratteristiche morfologiche e biochimiche dell'apoptosi. La risposta sembra essere la seguente. Quando il ligando di Fas si lega al suo recettore inizia una cascata di eventi che porta all'attivazione della caspasi. Il primo evento in questa cascata è l'aggregazione del complesso Fas/FasL. Il passaggio successivo prevede la formazione di una struttura composta da più proteine chiamata DISC e situata immediatamente al di sotto della superficie della membrana plasmatica. Componenti chiave di questo passaggio comprendono una proteina chiamata FADD che si lega alla coda citoplasmatica di Fas. FADD a sua volta interagisce con la procaspasi-8, evento che determina l'attivazione dell'enzima, la quale poi conduce all'attivazione della cascata mediata dalle caspasi e infine all'apoptosi. L'attivazione di queste vie implica l'esistenza di un interruttore molecolare che interrompe il processo quando ciò si rivela necessario. Nel caso in questione, l'interruttore che interrompe il processo è una proteina detta FLIP, la quale compete con la caspasi-8 per un sito di legame su FADD; ciò regola la velocità con cui la cellula va incontro ad apoptosi.
Morte cellulare nel sistema nervoso
La morte cellulare nel sistema nervoso è stata identificata da quasi un secolo. Nel 1949 Rita Levi-Montalcini dimostrò che nell'embrione di pollo un notevole numero di neuroni andava incontro a morte durante lo sviluppo del sistema nervoso. Questi primi studi aprirono la strada alla comprensione del ruolo svolto dalla morte cellulare nei processi ontogenetici. L'interesse per la morte cellulare nel sistema nervoso ebbe nuovo stimolo soltanto negli anni Settanta, quando furono presentati i risultati degli importanti lavori sperimentali di Andrew H. Wyllie e dei suoi colleghi.
Studi successivi dimostrarono che la morte cellulare, documentata precedentemente da Levi-Montalcini, era in realtà l'apoptosi. I neuroni morenti mostravano le classiche caratteristiche morfologiche dell'apoptosi e, inoltre, frammentavano il loro DNA in pezzi delle dimensioni di un nucleosoma. Il fatto che l'apoptosi delle cellule nervose fosse un processo attivo dipendente dalla sintesi di RNA e proteine venne dimostrato coltivando cellule neuronali in assenza di fattori di crescita, come per esempio quello delle fibre nervose (NGF, Nerve growth factor). In questo modo le cellule andavano incontro ad apoptosi che poteva essere prevenuta trattandole con un inibitore della sintesi proteica come la cicloesimmide. Attualmente è noto che durante l'ontogenesi viene prodotta un'enorme quantità di neuroni, i quali sopravvivono se possono formare connessioni con le cellule circostanti, altrimenti muoiono. Tali connessioni non soltanto rendono le cellule vitali, ma sono anche utili nella costruzione del sistema nervoso. Quindi, se non si connettono con i neuroni vicini, i neuroni neoformati muoiono; la formazione di connessioni è pertanto, per i neuroni, un incentivo alla sopravvivenza. Una delle caratteristiche delle malattie neurodegenerative degli esseri umani è la morte cellulare nel sistema nervoso centrale. Sebbene ancora si discuta sul fatto che la morte cellulare osservata avvenga oppure no per apoptosi, le prove a disposizione sono a favore dell'apoptosi. Con la perdita di cellule si verifica un deterioramento delle funzioni cerebrali che si manifesta in molti modi. Prevenendo la perdita di cellule cerebrali, è probabile che si possa intervenire efficacemente in malattie quali il morbo di Alzheimer e il morbo di Parkinson.
Bibliografia
Cohen, Duke 1984: Cohen, J. John - Duke, Rick C., Glucocorticoid activation of a calcium-dependent endonuclease in thymocyte nuclei leads to cell death, "Journal of immunology", 132, 1984, pp. 38-42.
Fadok, Chimini 2001: Fadok, Valerie A. - Chimini, Giovanna, The phagocytosis of apoptotic cells, "Seminars in immunology", 13, 2001, pp. 365-372.
Irmler 1997: Irmler, Martin e altri, Inhibition of death receptor signals by cellular FLIP, "Nature", 388, 1997, pp. 190-195.
Keeshan 2001: Keeshan, Karen e altri, Elevated Bcr-Abl expression levels are sufficient for a haematopoietic cell line to acquire a drug-resistant phenotype, "Leukemia", 15, 2001, pp. 1823-1833.
Kerr 1972: Kerr, John F.R. - Wyllie, Andrew H. - Currie, Alastair R., Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics, "British journal of cancer", 26, 1972, pp. 239-257.
Martin 1992: Martin, Diane P. e altri, Biochemical characterization of programmed cell death in NGF-deprived sympathetic neurons, "Journal of neurobiology", 23, 1992, pp. 1205-1220.
McDonnell, Korsmeyer 1991: McDonnell, Timothy I. - Korsmeyer, Stanley J., Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18), "Nature", 349, 1991, pp. 254-256.
Nagata, Golstein 1995: Nagata, Shigekazu - Golstein, Pierre, The Fas death factor, "Science", 267, 1995, pp. 1449-1456.
Oltvai 1993: Oltvai, Zoltán N. - Milliman, Curt L. - Korsmeyer, Stanley J., Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death, "Cell", 74, 1993, pp. 609-619.
Samali, Cotter 1996: Samali, Afshin - Cotter, Thomas G., Heat shock proteins increase resistance to apoptosis, "Experimental cell research", 223, 1996, pp. 163-170.
Yonish-Rouach 1991: Yonish-Rouach, Elisheva e altri,Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6, "Nature", 352, 1991, pp. 345-347.